Die polypoidale choroidale Vaskulopathie (PCV) ist eine Erkrankung, die durch ein abnormales verzweigtes Gefäßnetzwerk (BVN) der Aderhaut und polypoidale Gefäßerweiterungen an dessen Enden gekennzeichnet ist. 5) Es handelt sich um einen Subtyp der altersbedingten Makuladegeneration (AMD), der in der Indocyaningrün-Fluoreszenz-Angiographie (ICGA) polypoidale Erweiterungen der Aderhautgefäße zeigt, orange-rote kugelförmige Läsionen unter dem retinalen Pigmentepithel (RPE) bildet und eine seröse oder hämorrhagische Pigmentepithelabhebung (PED) verursacht.
Erstmals in den 1980er Jahren als „idiopathische hämorrhagische RPE-Abhebung“ beschrieben 5). Heute wird sie als eine Erkrankung des pachychoroidalen Spektrums betrachtet, das ein Kontinuum mit der zentralen serösen Chorioretinopathie (CSC) und der pachychoroidalen Neovaskularisation (PNV) bildet. 5, 6) Es wird darauf hingewiesen, dass sich das Krankheitskonzept in Zukunft weiter ändern könnte.
Tritt hauptsächlich bei Männern im Alter von 50–65 Jahren auf. In der gesamten asiatischen Bevölkerung macht sie 22–62 % der Patienten mit exsudativer nAMD aus, 5) und einige Berichte geben an, dass etwa die Hälfte der exsudativen AMD PCV ist. 11) Bei europäischen und amerikanischen Weißen liegt der Anteil bei nur etwa 10–20 %. 5) Es wird geschätzt, dass die Weltbevölkerung über 65 Jahre bis 2050 1,5 Milliarden übersteigen wird, was einen Anstieg der nAMD- und PCV-Patienten erwarten lässt. 5)
QIst die polypoidale choroidale Vaskulopathie (PCV) dieselbe Erkrankung wie die altersbedingte Makuladegeneration (AMD)?
A
Die polypoidale choroidale Vaskulopathie wird oft als Subtyp der nAMD klassifiziert, unterscheidet sich jedoch in Pathologie, Therapieansprechen und genetischem Hintergrund. Im Vergleich zur nAMD neigt sie zu mehr Blutungen und weniger IRF. Derzeit wird diskutiert, ob sie als eigenständige Krankheitsentität im pachychoroidalen Spektrum betrachtet werden sollte. 5, 6)
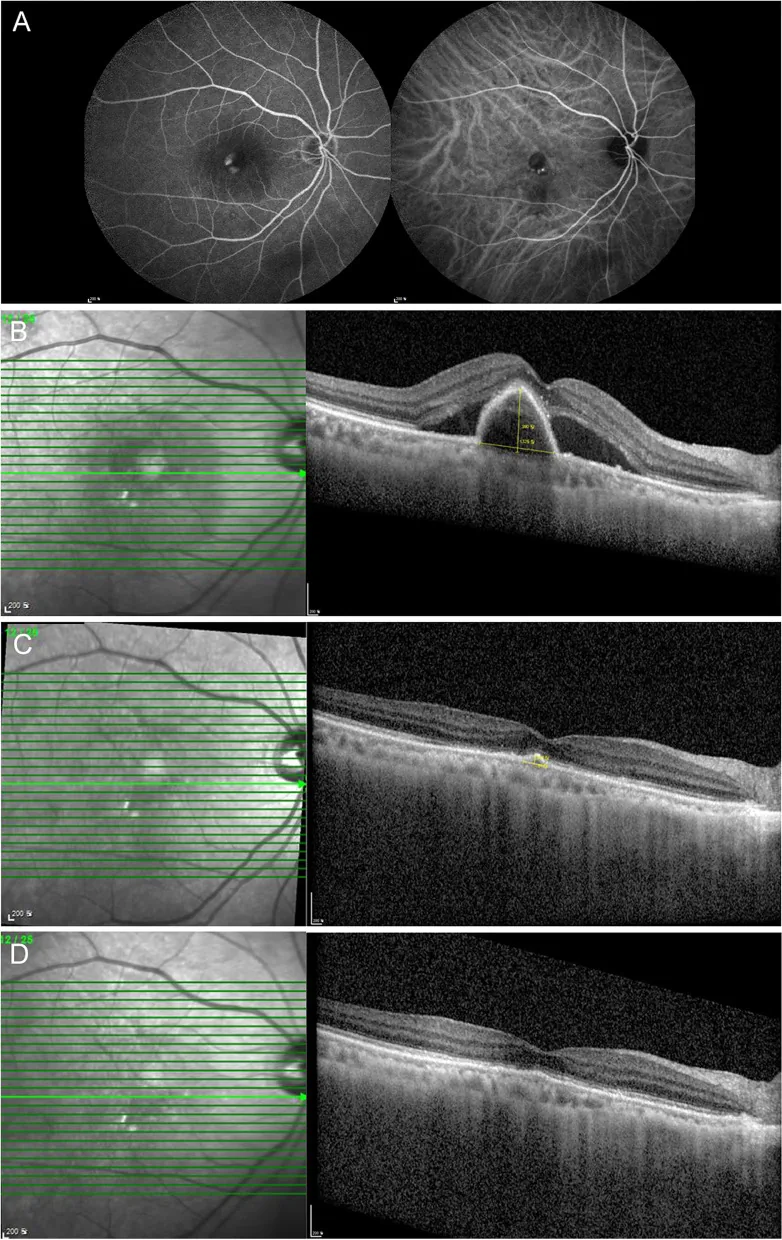
Nam SW, et al. Response to brolucizumab treatment for refractory serous pigment epithelial detachment secondary to polypoidal choroidal vasculopathy. BMC Ophthalmol. 2022. Figure 2. PMCID: PMC9749193. License: CC BY.
A zeigt eine Fluoreszenzangiographie und Indocyaningrün-Angiographie einer polypoidalen choroidalen Neovaskularisation, während B, C und D OCT-Bilder der Verbesserung der serösen Pigmentepithelabhebung (PED) und subretinalen Flüssigkeit (SRF) im Verlauf der Behandlung zeigen. Dies entspricht der serösen PED, die im Abschnitt „2. Hauptsymptome und klinische Befunde“ behandelt wird.
Sehverschlechterung: Läsionen, die die Fovea betreffen, führen zu einer akuten und fortschreitenden Sehverschlechterung. 5)
Metamorphopsie (Verzerrung): Aufgrund exsudativer Veränderungen der Makula erscheinen gerade Linien verbogen.
Zentralskotom: Wenn die Läsion das Zentrum der Makula erreicht, wird ein dunkler Bereich in der Mitte des Gesichtsfelds wahrgenommen.
Plötzliche Sehverschlechterung: Tritt bei massiver subretinaler Blutung (SMH) auf. Charakteristisch sind starke hämorrhagische Veränderungen; die 5-Jahres-Inzidenz von SMH wird mit etwa 10 % angegeben. 5)
Im Vergleich zur typischen nAMD zeigt die polypoidale choroidale Vaskulopathie reichlich Blutungen, während IRF (intraretinale Flüssigkeit) tendenziell geringer ist. Auch seröse PED und subretinale Blutungen sind häufig.
Klinische Befunde (vom Arzt bei der Untersuchung festgestellte Befunde)
Bei der Fundusuntersuchung sind orangefarbene erhabene Läsionen charakteristisch. Sie werden als polypoidale Gefäßerweiterungen unter dem RPE beobachtet. Häufig treten sie zusammen mit hämorrhagischer PED oder serös-hämorrhagischer PED auf.
Merkmale der PCV
Orangerote kugelförmige Läsion : polypoide Gefäßerweiterung unter dem RPE. Bei der Ophthalmoskopie als orangerote kugelförmige Läsion sichtbar.
Hämorrhagische Pigmentepithelabhebung : steile Vorwölbung durch massive Blutung unter dem RPE. Tendenz zu wenig intraretinaler Flüssigkeit.
Serös-hämorrhagische Pigmentepithelabhebung : RPE-Abhebung mit einer Mischung aus seröser Flüssigkeit und Blut.
Vergleich mit nAMD
PCV : viel Blutung, wenig intraretinale Flüssigkeit, Neigung zu submakulärer Blutung. ICGA erforderlich. Polypenverschluss ist das Behandlungsziel.
ICGA: Charakteristische polypoide Aderhautgefäßerweiterungen und ein abnormes Gefäßnetzwerk (Netzwerkgefäße) werden festgestellt. Der Nachweis von Polypenherden ist die definitive Diagnose. 5, 7)
Fluoreszein-Fluoreszenz-Fundusangiographie (FA): Die den Polypenherden entsprechenden Bereiche zeigen relativ früh eine Hyperfluoreszenz. Im Spätstadium zeigt sich häufig ein Window-Defekt entsprechend den atrophischen Herden.
Die OCTA kann Gefäße nicht-invasiv darstellen, und das BVN (abnormes Gefäßnetzwerk) wird oft deutlicher als mit der ICGA dargestellt. 5) Eine BNN-3-Typen-Klassifikation wurde berichtet, mit einer PCV-Erkennungssensitivität von 82,6 % und einer Spezifität von 100 %. 5) Allerdings hat die Erkennung von Polypenherden Grenzen und erreicht nicht die der ICGA. Für die Typdiagnose von PCV oder retinaler angiomatöser Proliferation (RAP) gibt es mit der alleinigen OCTA noch Herausforderungen, und die Diagnose mittels multimodaler Bildgebung ist wichtig.
Die polypoide Aderhautvaskulopathie ist eine spezielle Form der Typ-1-Aderhautneovaskularisation (CNV) unter dem RPE. 5, 8) Das grundlegende pathologische Substrat ist das Pachychoroid, ein Zustand, der durch eine Erweiterung der Haller-Schicht (äußere Aderhautgefäßschicht) und eine damit verbundene Ausdünnung der Sattler-Schicht und der Choriokapillaris gekennzeichnet ist. 5)
Vortexvenenstauung: Eine Stauung der Aderhautvenen führt zu einer Erweiterung der Haller-Schicht. Eine Anastomose der erweiterten Pachychoroid-Gefäße mit den Vortexvenen wird in etwa 90 % der Fälle beobachtet. 5)
Ischämie/Verschluss der Choriokapillaris: Eine Ischämie auf der Ebene der Choriokapillaris induziert die Bildung des Neovaskularisationskomplexes (BVN). 5)
Schädigung von RPE und Bruch-Membran: Die chronische Aderhautstauung schädigt das RPE und die Bruch-Membran, was unter Begleitung einer erhöhten Proteaseaktivität zur Bildung von Polypenherden führt. 5)
Es wurden Assoziationen mit Polymorphismen der Gene CFH (Komplementfaktor H) und ARMS2/HTRA1 berichtet. 5) Es gibt einige gemeinsame Suszeptibilitätsgene mit der typischen nAMD, aber beim nicht-pachychoroidalen Typ (drusen-getriebene PCV, normale Aderhautdicke) ist die Assoziation mit ARMS2/HTRA1 stärker. 5, 8) Auch SNPs des ANGPT2-Gens und Missense-Mutationen des FGD6-Gens wurden als mit der PCV-Entstehung assoziiert vermutet. 5)
Pachychoroidaler Typ und nicht-pachychoroidaler Typ
Etwa die Hälfte der PCV-Augen zeigt eine normale Aderhautdicke. 5) Der pachychoroidale Typ hat eine Tendenz zu jüngerem Alter, weist viele CSC-ähnliche Merkmale auf, geht mit einer Aderhautgefäß-Hyperpermeabilität einher und kann eine Anti-VEGF-Resistenz zeigen. Der nicht-pachychoroidale Typ ist drusen-getrieben (AMD-ähnliche Merkmale) und hat eine stärkere Assoziation mit ARMS2/HTRA1. 5, 8)
Durchblutungsdefizite der Choriokapillaris (CCFD) sind nicht nur in PCV-erkrankten Augen, sondern auch in gesunden Partneraugen erhöht, was darauf hindeutet, dass Pachychoroid eher als systemische Prädisposition denn als lokaler Faktor wirken könnte. 5)
Sekundäre PCV nach Chorioiditis: Chronische Schädigung des RPE und der Bruch-Membran durch tuberkulöse Chorioiditis kann als Grundlage dienen. Es wurde ein Fall berichtet, bei dem etwa 20 Jahre nach der Entzündung eine PCV auftrat und die Sehschärfe nach drei Injektionen von Aflibercept von 6/9 auf 6/6 verbessert wurde. 1)
Rasche Progression nach COVID-19-Impfung: Ein Fall (79-jähriger Mann) wurde berichtet, bei dem 16 Stunden nach der dritten Dosis Symptome auftraten und innerhalb von zwei Wochen eine rasche Progression zu einer massiven SMH erfolgte. 4)
Die von der EVEREST-Studiengruppe festgelegten ICGA-basierten Diagnosekriterien werden international weitgehend verwendet. 7, 10) Die Diagnose einer PCV wird gestellt, wenn eines der folgenden Kriterien erfüllt ist.
Nachweis von orange-roten kugelförmigen Läsionen in der Fundusuntersuchung
Nachweis von knotigen hyperfluoreszenten Läsionen (Polypen) in der ICGA
Nachweis eines abnormalen verzweigten Gefäßnetzwerks (BVN) im ICGA
Die ICGA ist der Goldstandard für die Diagnose der PCV. 5, 7) Sie eignet sich hervorragend zur Darstellung der Aderhautgefäße und kann aufgrund der langen Wellenlänge des ICG Gefäßstrukturen auch durch Blut, Flüssigkeit oder Lipide unter dem RPE sichtbar machen. 5) Sie identifiziert frühe hyperfluoreszierende polypoidale Läsionen und das BVN.
Für die Unterscheidung der PCV allein durch OCT wurde ein AUC von 0,90 nach APOIS-Kriterien (APOIS PCV Workgroup) berichtet. 5, 12) Das Double-Layer-Zeichen ist für das Screening in Einrichtungen ohne ICGA nützlich, aber die Sensitivität beträgt nur 59 %. 5) Wenn ICGA nicht verfügbar ist, kann die Kombination von OCT und OCTA eine Alternative sein, aber für die Therapieentscheidung (z. B. Hinzufügen von PDT) ist ICGA weiterhin wichtig.
Typische AMD (Typ-1/2-CNV) : Das Vorhandensein oder Fehlen von polypoiden Läsionen in der ICGA ist der entscheidende Punkt für die Differenzierung.
Zentrale seröse Chorioretinopathie (CSC) : Eine Differenzierung innerhalb des pachychoroidalen Spektrums ist erforderlich. Die Aderhautdicke und das Vorhandensein von Neovaskularisationen sind hilfreich.
Retinale Mikroaneurysmen : Abgrenzung zur hämorrhagischen PED erforderlich.
QKann eine PCV ohne ICGA diagnostiziert werden?
A
Fortschritte in OCT und OCTA ermöglichen zunehmend eine ICGA-unabhängige Diagnoseunterstützung, aber derzeit ist ICGA der Goldstandard. Obwohl die APOIS-Kriterien mit einem AUC von 0,90 eine hohe Unterscheidungsfähigkeit zeigen, bleibt ICGA für die Therapieentscheidung (z. B. Hinzufügen von PDT) ein wichtiger Test. 5, 12)
Gemäß den Behandlungsleitlinien der Arbeitsgruppe des Ministeriums für Gesundheit, Arbeit und Soziales von 2012 wird die Wahl der Behandlung basierend auf der Sehschärfe empfohlen.
Fälle mit guter Sehschärfe (0,6 oder besser) : Erwägung einer Anti-VEGF-Monotherapie.
Bei PCV hängt die Rückbildung polypoider Läsionen mit dem Rezidiv nach der Behandlung zusammen. Mit Ranibizumab betrug die vollständige Polypenrückbildungsrate 20–30 %, mit Aflibercept dagegen 40–50 %, sodass in den letzten Jahren unabhängig von der Sehschärfe vermehrt VEGF-Inhibitoren allein eingesetzt werden. 9)
Die intravitreale Injektion von Anti-VEGF-Medikamenten ist die Erstlinientherapie der PCV. 5, 9)
Aflibercept zeigt eine höhere Polypenverschlussrate als Ranibizumab. In der PLANET-Studie wurde die Nichtunterlegenheit von Aflibercept allein gegenüber Aflibercept + PDT nachgewiesen (1-Jahres-Ergebnisse). 9) Die Polypenverschlussrate erreichte auch in der Monotherapiegruppe über 85 %. 3, 9)
Vella et al. (2021) berichteten, dass bei PCV-Fällen, die auf 6 Gaben Ranibizumab nicht ansprachen, eine einzige Gabe von Aflibercept zu einer vollständigen Resorption der subretinalen Flüssigkeit (SRF) führte. 3)
Bestrahlungsgröße: maximaler Läsionsdurchmesser basierend auf Kontrastmittelbefunden + 1000 μm
Vermeidung direkter Sonneneinstrahlung für 2 Tage nach der Behandlung erforderlich
Bei Kombination von PDT + VEGF-Inhibitor kann der VEGF-Inhibitor vor der PDT (innerhalb einer Woche) oder am selben Tag der PDT (unter Lichtschutz) verabreicht werden.
In der EVEREST-I-Studie betrug die Läsionsverschlussrate bei PDT + Ranibizumab-Kombination oder PDT allein 77,8 %, während sie bei Ranibizumab allein nur 26,7 % betrug.10) In der EVEREST-II-Studie (RCT) zeigte die Gruppe mit PDT + Ranibizumab nach 24 Wochen eine um 9,6 Buchstaben bessere Sehschärfenverbesserung als die Gruppe mit Ranibizumab allein.10)
Als minimalinvasive Alternative zur PDT wurde die Wirksamkeit des 577-nm-Mikropulslasers berichtet.
Jafar et al. (2024) berichteten über einen Fall von PCV, der mit einem 577-nm-Mikropulslaser (Einschaltdauer 5 %, 400 mW, 200 μm, 200 ms) behandelt wurde, mit vollständigem Verschwinden der subretinalen Flüssigkeit nach 12 Wochen. Die Sehschärfe verbesserte sich von 20/60 auf 20/25.2)
Bei Fällen mit massiver submakulärer Blutung kann eine Vitrektomie zur Hämatomentfernung erforderlich sein.5)
Sasajima et al. (2022) führten bei einem schnell fortschreitenden PCV (79-jähriger Mann) nach der dritten COVID-19-Impfung eine Vitrektomie mit Gewebe-Plasminogen-Aktivator (tPA 12,5 μg/0,05 mL) + SF6-Gas 1,2 mL durch. Am 13. postoperativen Tag wurde eine Abnahme von SMH und SRF festgestellt. 4)
QWelches soll man wählen: Aflibercept oder Ranibizumab?
A
Aflibercept hat eine vollständige Polypenrückbildungsrate von 40–50 %, höher als Ranibizumab (20–30 %). Die PLANET-Studie hat auch die Nichtunterlegenheit von Aflibercept-Monotherapie gegenüber PDT gezeigt, und es wird derzeit als Anti-VEGF-Medikament der ersten Wahl für PCV empfohlen. 3, 9)
PCV ist eine repräsentative Erkrankung des Pachychoroid-Spektrums (PSD). 5, 6) Zum PSD gehören die Pachychoroid-Pigmentepitheliopathie, die Pachychoroid-Neovaskularisation (PNV), die zentrale seröse Chorioretinopathie (CSC) und das peripapilläre Pachychoroid-Syndrom (PPS). 5) In letzter Zeit wurde die periphere exsudative hämorrhagische Chorioretinopathie (PEHCR) zum Pachychoroid-Spektrum hinzugefügt und als peripherer Phänotyp des PCV betrachtet. 5, 13) PCV ist eine spezielle Form der Typ-1-CNV unter dem RPE, bei der auf der Grundlage einer Ischämie der Choriokapillaris ein abnormales Gefäßnetzwerk entsteht.
Die erweiterten großkalibrigen Aderhautgefäße (Pachygefäße) in der Haller-Schicht stehen im Zentrum der Pathologie. 5) Anastomosen zwischen den oberen und unteren Vortexvenen werden bei etwa 90 % der PSD-Augen gefunden, was zum Verschwinden der horizontalen Wasserscheidenzone führt. 5) Diese Vortexvenenstauung verursacht schrittweise Veränderungen: Erweiterung der Haller-Schicht → Verdünnung der Sattler-Schicht und der Choriokapillaris → Ischämie auf Kapillarebene → Bildung eines neovaskulären Komplexes. 5)
Chronische Aderhautstauung führt zu folgenden schrittweisen Veränderungen.
Stoffwechselstörung und Funktionsstörung des RPE
Strukturelle Zerstörung der Bruch-Membran durch erhöhte Proteaseaktivität5)
Bildung eines abnormalen verzweigten Gefäßnetzwerks (BVN)
Auftreten von polypoiden erweiterten Läsionen
Die Aderhaut besitzt keinen Kapillarkomplex wie die Netzhaut und ist direkt dem pulsierenden Blutfluss ausgesetzt. Daher neigt die Spitze der CNV zur Erweiterung und Bildung von Polypen.5) Zudem ist der choroidale Kapillarplatten-Perfusionsdefekt (CCFD) nicht nur in PCV-Augen, sondern auch in gesunden Partneraugen erhöht, was darauf hindeutet, dass die Pachychoroidea als bilaterale systemische Prädisposition wirken könnte.5)
Der pachychoroide PCV-Typ tritt häufiger bei jüngeren Patienten auf, zeigt viele CSC-ähnliche Merkmale, ist mit choroidaler vaskulärer Hyperpermeabilität (CVH) verbunden und kann eine Anti-VEGF-Resistenz aufweisen.5, 8) Der nicht-pachychoroide Typ (drusen-getriebene PCV) hingegen zeigt AMD-ähnliche Merkmale und ist stark mit ARMS2/HTRA1-Risikoallelen assoziiert.5, 8) Da beide Typen unterschiedliche klinische Verläufe und Therapieansprechen zeigen, ist eine Individualisierung der Behandlungsstrategie erforderlich.
Direkte Schädigung von RPE und Bruch-Membran durch Entzündung kann den Boden für die PCV-Entwicklung bereiten.1) In einem Fall, bei dem 20 Jahre nach einer tuberkulösen Chorioiditis eine PCV auftrat, wurde die Akkumulation von Gewebeschäden durch chronische Entzündung als ursächlich für die PCV-Entwicklung vermutet.1)
7. Aktuelle Forschung und Zukunftsperspektiven (Forschungsstadium)
Brolucizumab (ein hochaffiner Einzelkettenantikörper gegen VEGF-A) zeigte eine nicht unterlegene Visusverbesserung und bessere anatomische Ergebnisse im Vergleich zu bestehenden Anti-VEGF-Medikamenten. 5) Zusammen mit Faricimab (VEGF-A/Ang-2-Dualinhibition) gilt es als Kandidat für die Verlängerung der Behandlungsintervalle (Treat-and-Extend; T&E). 6) Beide warten auf die Akkumulation von Langzeitdaten.
Das PDS, ein Gerät zur kontinuierlichen Freisetzung von Anti-VEGF-Medikamenten, wird als Alternative zu häufigen intravitrealen Injektionen entwickelt. 5) Es könnte zur Verringerung der Behandlungslast bei nAMD-Patienten, einschließlich PCV, beitragen.
T&E ist ein Schema, bei dem nach anfänglicher Fixdosis die Behandlungsintervalle je nach individuellem Rezidivrisiko verlängert werden. 6) Seine Anwendung bei PCV-Patienten wird diskutiert, aber das optimale Protokoll ist noch nicht etabliert.
Eine KI-Diagnose basierend auf OCT-Befunden (APOIS-Kriterien) zeigte eine Unterscheidungsfähigkeit von nAMD-PCV mit einer AUC von 0,90. 5, 12) In der TIGER-Studie wird ein neues System zur Beurteilung von Diagnose und Therapieansprechen unter Verwendung von OCTA und KI untersucht. 5)
Mit der Verbreitung des Pachychoroid-Spektrum-Konzepts wird weiterhin diskutiert, ob PCV als Subtyp der nAMD oder als eigenständige Erkrankung betrachtet werden sollte. 6, 8) Pachychoroide und nicht-pachychoroide (drusen-getriebene) PCV unterscheiden sich in Aderhautdicke, Therapieansprechen und Langzeitprognose, 14) und die Verfeinerung der Behandlungsstrategie, die nach Unterscheidung beider Typen individuell über Anti-VEGF allein oder in Kombination mit PDT entscheidet, ist eine zukünftige Hauptaufgabe.
Die Prognose der PCV ist eng mit dem Grad der Regression der polypoiden Läsionen verbunden. Die vollständige Polypenregressionsrate unter Aflibercept beträgt 40–50 %, höher als die 20–30 % unter Ranibizumab. 9) Bei vollständiger Regression sinkt das Rezidivrisiko, bei unvollständiger Regression kommt es jedoch häufig zu erneuter Flüssigkeitsansammlung, sodass regelmäßige Bildgebung unerlässlich ist.
Auch nach erfolgreicher Behandlung und Verschwinden der exsudativen Veränderungen kann es durch Polypenruptur zu einer plötzlichen massiven subretinalen Blutung (SMH) kommen. Die Rate der SMH über 5 Jahre wird mit etwa 10 % angegeben, 5) und bei plötzlichen Sehstörungen sollte wiederholt zur sofortigen Konsultation geraten werden. Bei massiven subfovealen Blutungen ist die Sehprognose schlecht, und ein chirurgischer Eingriff wie tPA + Gas-Austausch kann erforderlich sein.
Die pachychoroide PCV neigt zur Resistenz gegen Anti-VEGF und erfordert häufig eine kombinierte PDT. 5, 8) Die nicht-pachychoroide (drusen-getriebene) Form hingegen zeigt einen AMD-ähnlichen chronischen Verlauf und erfordert eine langfristige Erhaltungstherapie. 5, 8) Beide Subtypen haben eine hohe Rezidivrate, und regelmäßige OCT- und ICGA-Kontrollen sind auch nach Behandlungsende wichtig.
Mohankumar A, Mohan S, Rajan M. Polypoidal choroidal vasculopathy 20 years after resolution of tubercular choroiditis. Digital journal of ophthalmology : DJO. 2023;29(3):94-96. doi:10.5693/djo.02.2023.07.003. PMID:37780035; PMCID:PMC10539005.
Jafar SM, Hussein ZR, Yasir MB. A case of treating polypoidal choroidal vasculopathy subretinal fluid by subthreshold micropulse laser. American journal of ophthalmology case reports. 2024;36:102225. doi:10.1016/j.ajoc.2024.102225. PMID:39691633; PMCID:PMC11650130.
Vella G, Sacconi R, Borrelli E, Bandello F, Querques G.. Polypoidal choroidal vasculopathy in a patient with early-onset large colloid drusen. Am J Ophthalmol Case Rep. 2021;22:101085. doi:10.1016/j.ajoc.2021.101085. PMID:33898862; PMCID:PMC8056241.
Sasajima H, Zako M, Maeda R, Ueta Y. Rapid Progression of Polypoidal Choroidal Vasculopathy following Third BNT162b2 mRNA Vaccination. Case reports in ophthalmology. 2022;13(2):459-464. doi:10.1159/000525151. PMID:35950020; PMCID:PMC9247562.
Sen P, Manayath G, Shroff D, Salloju V, Dhar P. Polypoidal Choroidal Vasculopathy: An Update on Diagnosis and Treatment. Clinical ophthalmology (Auckland, N.Z.). 2023;17:53-70. doi:10.2147/OPTH.S385827. PMID:36636621; PMCID:PMC9831529.
Cheung CMG, Dansingani KK, Koizumi H, et al. Pachychoroid disease: review and update. Eye (Lond). 2025;39(5):819-834. doi:10.1038/s41433-024-03253-4.
Koh AH, Expert PCV Panel, Chen LJ, Chen SJ, Chen Y, Giridhar A, Iida T, Kim H, Yuk Yau Lai T, Lee WK, Li X, Han Lim T, Ruamviboonsuk P, Sharma T, Tang S, Yuzawa M. Polypoidal choroidal vasculopathy: evidence-based guidelines for clinical diagnosis and treatment. Retina. 2013;33(4):686-716. doi:10.1097/iae.0b013e3182852446. PMID:23455233.
Cheung CMG, Lai TYY, Ruamviboonsuk P, Chen SJ, Chen Y, Freund KB, Gomi F, Koh AH, Lee WK, Wong TY.. Polypoidal Choroidal Vasculopathy: Definition, Pathogenesis, Diagnosis, and Management. Ophthalmology. 2018;125(5):708-724. doi:10.1016/j.ophtha.2017.11.019. PMID:29331556.
Lee WK, Iida T, Ogura Y, Chen SJ, Wong TY, Mitchell P, et al. Efficacy and Safety of Intravitreal Aflibercept for Polypoidal Choroidal Vasculopathy in the PLANET Study: A Randomized Clinical Trial. JAMA ophthalmology. 2018;136(7):786-793. doi:10.1001/jamaophthalmol.2018.1804. PMID:29801063; PMCID:PMC6136040.
Lim TH, Lai TYY, Takahashi K, Wong TY, Chen LJ, Ruamviboonsuk P, Tan CS, Lee WK, Cheung CMG, Ngah NF, Patalauskaite R, Margaron P, Koh A, EVEREST II Study Group.. Comparison of Ranibizumab With or Without Verteporfin Photodynamic Therapy for Polypoidal Choroidal Vasculopathy: The EVEREST II Randomized Clinical Trial. JAMA Ophthalmol. 2020;138(9):935-942. doi:10.1001/jamaophthalmol.2020.2443. PMID:32672800; PMCID:PMC7366282.
Voraporn Chaikitmongkol, Chui Ming Gemmy Cheung, Hideki Koizumi, Vishal Govindahar, Jay Chhablani, Timothy Y.Y. Lai. Latest Developments in Polypoidal Choroidal Vasculopathy: Epidemiology, Etiology, Diagnosis, and Treatment. Asia-Pacific Journal of Ophthalmology. 2020;9(3):260-268. doi:10.1097/01.apo.0000656992.00746.48.
Chong Teo KY, Sadda SR, Gemmy Cheung CM, Chakravarthy U, Staurenghi G, Invernizzi A, et al. Non-ICGA treatment criteria for Suboptimal Anti-VEGF Response for Polypoidal Choroidal Vasculopathy: APOIS PCV Workgroup Report 2. Ophthalmology. Retina. 2021;5(10):945-953. doi:10.1016/j.oret.2021.04.002. PMID:33866022.
Dansingani KK, Balaratnasingam C, Nishi K, et al. Understanding aneurysmal type 1 neovascularization (polypoidal choroidal vasculopathy): a lesson in the taxonomy of ‘expanded spectra’ — a review. Prog Retin Eye Res. 2018;65:89-117.
Miyake M, Ooto S, Yamashiro K, Takahashi A, Yoshikawa M, Akagi-Kurashige Y, Ueda-Arakawa N, Oishi A, Nakanishi H, Tamura H, Tsujikawa A, Yoshimura N.. Pachychoroid neovasculopathy and age-related macular degeneration. Sci Rep. 2015;5:16204. doi:10.1038/srep16204. PMID:26542071; PMCID:PMC4635432.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.