Cryptophthalmos (ตาถูกซ่อน) เป็นความผิดปกติแต่กำเนิดที่เปลือกตาหลอมรวมปกคลุมลูกตาและไม่มีรอยแยกของเปลือกตา
แบ่งออกเป็น 3 ประเภท: แบบสมบูรณ์ แบบไม่สมบูรณ์ และแบบไม่ครบถ้วน
Cryptophthalmos พบได้ใน 80-93% ของผู้ป่วยกลุ่มอาการ Fraser สาเหตุมักเกิดจากการถ่ายทอดทางพันธุกรรมแบบออโตโซมด้อยจากการกลายพันธุ์ของยีน FRAS1 และ FREM2
อุบัติการณ์พบได้น้อยมาก คือ 0.043 ต่อทารกเกิดมีชีพ 10,000 คน และมีรายงานในเอกสารเพียง 55 ราย ณ ปี 2018
เกือบทุกกรณีไม่มีการมองเห็น หรือมองเห็นได้น้อยมาก การผ่าตัดมีเป้าหมายหลักเพื่อความสวยงามและการสร้างบริเวณดวงตาขึ้นใหม่
สามารถตรวจพบได้ด้วยอัลตราซาวนด์ก่อนคลอดประมาณอายุครรภ์ 18 สัปดาห์ โดยพบว่าไม่มีรอยแยกของเปลือกตา
Cryptophthalmos มาจากภาษาละติน แปลว่า “ตาที่ถูกซ่อน” หมายถึงภาวะที่ผิวหนังเปลือกตาที่เชื่อมติดกันปกคลุมลูกตาและเบ้าตา โดยไม่มีรอยแยกของเปลือกตา ผิวหนังหน้าผากต่อเนื่องกับผิวหนังแก้ม
อุบัติการณ์รายงานอยู่ที่ 0.043 ต่อทารกเกิดมีชีพ 10,000 คน และ 1.1 ต่อทารกตายคลอด 10,000 คน1) ณ ปี 2018 มีรายงานเพียง 55 รายในเอกสาร ความชุกโดยรวมของความผิดปกติของเปลือกตาคือ 0.06% โดย 2/3 ของกรณีเป็นแบบประปราย และ 1/3 มีปัจจัยทางพันธุกรรม1)
Cryptophthalmos จำแนกตามลักษณะทางสัณฐานวิทยาเป็น 3 ประเภท
ชนิดสมบูรณ์ (แบบฉบับ) : การอุดตันของเบ้าตา อย่างสมบูรณ์ ชนิดรุนแรงที่สุด ร่วมกับการไม่มีคิ้ว ขนตา และโครงสร้างต่อมชนิดไม่สมบูรณ์ (ไม่ปกติ) : มีหนังตาที่เป็นร่องรอยเหลืออยู่ มีถุงเยื่อบุตา ขนาดเล็กทางด้านข้างชนิดแท้ง (abortive form / ภาวะหนังตาติดลูกตา ตั้งแต่กำเนิด) : ไม่มีหนังตาบน ผิวหนังหน้าผากติดกับส่วนบนของกระจกตา
ทุกประเภทสามารถเป็นข้างเดียวหรือสองข้าง เกิดเดี่ยวหรือเป็นส่วนหนึ่งของกลุ่มอาการ ตาที่ซ่อนคือตาที่ถูกปกคลุมด้วยผิวหนังหนังตาที่เชื่อมติดกัน อาจเกิดเดี่ยวหรือเป็นหนึ่งในอาการของกลุ่มอาการเฟรเซอร์ หากขอบหนังตาไม่ถูกสร้างขึ้นเลย จะวินิจฉัยเป็นภาวะไม่มีหนังตาหรือตาที่ซ่อน
มีความสัมพันธ์อย่างใกล้ชิดกับกลุ่มอาการเฟรเซอร์ (Fraser syndrome) และส่วนใหญ่เป็นกรรมพันธุ์แบบออโตโซมัลด้อยจากการกลายพันธุ์ของยีน FRAS1, GRIP1 และ FREM2 พบภาวะตาซ่อน (cryptophthalmos) ใน 80-93% ของผู้ป่วยกลุ่มอาการเฟรเซอร์
Q
เด็กที่มีภาวะตาซ่อนสามารถฟื้นฟูการมองเห็นได้หรือไม่?
A
ในเกือบทุกกรณี ไม่มีการมองเห็น หรือแทบไม่มีเลย จุดประสงค์ของการผ่าตัดหลักคือเพื่อปรับปรุงรูปลักษณ์ด้านความสวยงามและสร้างบริเวณดวงตาขึ้นใหม่ การฟื้นฟูการมองเห็น นั้นเกิดขึ้นได้น้อยมาก ดูรายละเอียดในหัวข้อ “วิธีการรักษามาตรฐาน”
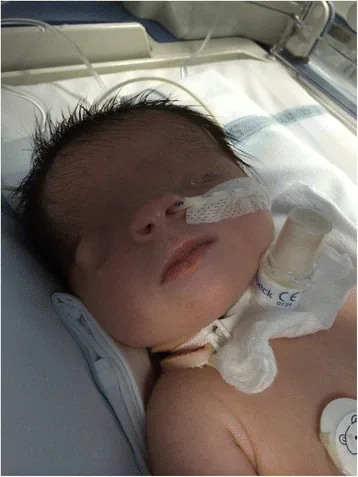
De Bernardo G, Giordano M, Di Toro A, et al. Prenatal diagnosis of Fraser syndrome: a matter of life or death? Ital J Pediatr. 2015 Nov 9;41:86. Figure 1. PMCID: PMC4640198. License: CC BY.
ภาพถ่ายใบหน้าทันทีหลังคลอด แสดงให้เห็นว่าไม่มีรอยแยกของเปลือกตาและผิวหนังปกคลุมผิวลูกตา ตาซ้ายมีขนาดเล็ก และตาขวาก็มีความผิดปกติของรูปร่าง ช่วยให้เข้าใจลักษณะภายนอกของตาซ่อน
ตาบอด : อาการที่รุนแรงที่สุด ดวงตาถูกผิวหนังปิดกั้น ไม่สามารถรับแสงจากภายนอกได้ตาบอดทั้งสองข้าง : ผู้ป่วยส่วนใหญ่เป็นตาบอดทั้งสองข้าง มีรายงานผู้ป่วยตาบอดข้างเดียวจำนวนน้อยไม่สมมาตร : โดยปกติจะสมมาตรในตาทั้งสองข้าง แต่หนึ่งในสามของผู้ป่วยมีลักษณะไม่สมมาตร
ผลการตรวจทางคลินิกหลักตามประเภทมีดังนี้
ชนิดสมบูรณ์
การอุดตันของเบ้าตา : ผิวหนังหน้าผากและแก้มต่อเนื่องกัน รอยแยกเปลือกตาหายไปโดยสิ้นเชิง
การไม่มีอวัยวะประกอบ : ไม่มีคิ้ว ขนตา และโครงสร้างต่อม
การร่วมกับลูกตาขนาดเล็ก : ผิวหนังชั้นผิวหลอมรวมกับกระจกตา ไม่มีถุงเยื่อบุตา ลูกตาขนาดเล็กพบได้บ่อยมาก อาจมีถุงน้ำในเบ้าตา
ชนิดไม่สมบูรณ์และชนิดไม่ครบ
ชนิดไม่สมบูรณ์ : เปลือกตาที่เป็นร่องรอยหลงเหลืออยู่ ความยาวรอยแยกเปลือกตาประมาณ 1/3 ของปกติ มีถุงเยื่อบุตา ขนาดเล็กทางด้านนอก ลูกตามีขนาดเล็กและเกือบถูกผิวหนังปกคลุม
ชนิดไม่สมบูรณ์ : ไม่มีหนังตาบน ผิวหนังหน้าผากติดกับกระจกตา ส่วนบน 75% กระจกตา ที่ถูกปกคลุมจะกลายเป็นเคราตินและขุ่น แต่กระจกตา ที่เปิดอยู่อาจใสได้
ในรายงานผู้ป่วย ทารกเพศชายอายุครรภ์ 39 สัปดาห์พบ cryptophthalmos ทั้งสองข้าง ไม่มีคิ้ว แนวเส้นผมหน้าผากผิดปกติ ปลายจมูกแหว่ง ใบหูต่ำ ตาห่างกัน และความผิดปกติของทวารหนักและทวารหนักส่วนล่าง 1) คลำพบถุงน้ำในเบ้าตา ที่เคลื่อนที่ได้ขนาด 1×1 ซม. ที่ตาขวา และตาซ้ายถูกฝังลึก 1) CT scan พบรูปแบบรอยนูนของสมองผิดปกติที่บริเวณท้ายทอยและตาซ้ายมีขนาดเล็กลง 1)
สาเหตุของ cryptophthalmos คือการกลายพันธุ์ของยีนในโปรตีนที่ประกอบเป็น FRAS/FREM complex
FRAS/FREM complex : โปรตีน FRAS1, FREM1 และ FREM2 รักษาการยึดเกาะระหว่างเยื่อฐานและเยื่อบุผิวระหว่างการพัฒนาของตัวอ่อน การกลายพันธุ์ทำให้การยึดเกาะบกพร่อง ส่งผลให้หนังตาแยกไม่สำเร็จ 1) Fraser syndrome (FRASRS1/2) : การถ่ายทอดทางพันธุกรรมแบบ autosomal recessive จากการกลายพันธุ์ของยีน FRAS1 หรือ FREM2 1) กลุ่มอาการ MOTA : เกิดจากการกลายพันธุ์ของยีน FREM1 ร่วมกับภาวะปากแหว่งเพดานโหว่ ความผิดปกติของทวารหนักและทวารหนัก ไตไม่เจริญ และอื่นๆ 1) .ตาที่ซ่อนอยู่ แบบแยกเดี่ยวตาที่ซ่อนอยู่ แบบแยกเดี่ยวข้างเดียวหรือทั้งสองข้างได้1) .รูปแบบการถ่ายทอดทางพันธุกรรม : มักเป็นแบบ autosomal recessive นอกจากนี้ยังมีรายงานแบบ autosomal dominant ด้วย
กลไกที่ทราบกันดีของการเกิดภาวะหนังตาบกพร่องแต่กำเนิด ได้แก่ การปิดรอยแยกของใบหน้าไม่สมบูรณ์ในระยะตัวอ่อน และการกดทับจากสายน้ำคร่ำ
การให้คำปรึกษาทางพันธุกรรม
ตาที่ซ่อนอยู่ อาจเกี่ยวข้องกับภาวะทางพันธุกรรม เช่น กลุ่มอาการเฟรเซอร์ แนะนำให้ปรึกษาผู้เชี่ยวชาญด้านพันธุศาสตร์เพื่อประเมินความเสี่ยงในการเกิดซ้ำ การตรวจแผง FRAS/FREM มีประโยชน์ในการวินิจฉัย
Q
Cryptophthalmos เกี่ยวข้องกับ Fraser syndrome อย่างไร?
A
Cryptophthalmos พบได้ใน 80-93% ของผู้ป่วย Fraser syndrome และการกลายพันธุ์ของยีน FRAS1/FREM2 เป็นสาเหตุหลัก คอมเพล็กซ์ FRAS/FREM มีความจำเป็นต่อการยึดเกาะระหว่างเยื่อฐานและเยื่อบุผิวในระหว่างการพัฒนาของตัวอ่อน และการกลายพันธุ์ทำให้เกิดความล้มเหลวในการแยกเปลือกตา 1) อย่างไรก็ตาม การกลายพันธุ์ของ FREM2 ก็สามารถทำให้เกิด cryptophthalmos แบบเดี่ยวได้เช่นกัน
สามารถตรวจพบได้ด้วยอัลตราซาวนด์ก่อนคลอดประมาณอายุครรภ์ 18 สัปดาห์ ผลการตรวจพบว่าไม่มีรอยแยกของเปลือกตาและผิวหนังต่อเนื่องกันจากหน้าผากถึงแก้ม หากพบนิ้วติดกัน, การสะท้อนเสียงของปอดเพิ่มขึ้น, และน้ำคร่ำน้อย มีความเป็นไปได้สูงว่าเป็น Fraser syndrome
เกณฑ์การวินิจฉัยกลุ่มอาการเฟรเซอร์แสดงไว้ด้านล่างนี้
การจำแนก รายการ เกณฑ์หลัก ตาซ่อน, นิ้วติดกัน, ความผิดปกติของอวัยวะสืบพันธุ์, ความผิดปกติของแขนขา เกณฑ์รอง ความผิดปกติของหูและจมูก, ปากแหว่งเพดานโหว่, ความผิดปกติของแนวเส้นผม, ความผิดปกติของไต
การตรวจที่จำเป็น:
การตรวจวัดสายตา ตาเหล่ และจอประสาทตา การมองเห็น การทดสอบการดึงรั้งภายใต้การดมยาสลบ : ยืนยันการมีอยู่ของเส้นใยที่ซ่อนอยู่CT/MRI : ประเมินรูปร่างของลูกตาและสมอง มีประโยชน์ในการยืนยันภาวะแทรกซ้อนของระบบประสาทส่วนกลาง เช่น รูปแบบรอยหยักของสมองที่ผิดปกติบริเวณท้ายทอย1)
การตรวจชุด FRAS/FREM มีความสำคัญในการยืนยันการวินิจฉัย1) เนื่องจากฟีโนไทป์ของกลุ่มอาการเฟรเซอร์และกลุ่มอาการ MOTA ทับซ้อนกัน การแยกความแตกต่างโดยอาศัยเฉพาะอาการทางคลินิกโดยไม่มีการตรวจทางพันธุกรรมอาจทำได้ยาก
การวินิจฉัยแยกโรครวมถึงภาวะไม่มีเปลือกตาแต่กำเนิด (ในกรณีที่บกพร่องบางส่วน), ภาวะตาเดียว, และภาวะตาสองข้างไม่เท่ากัน
เป้าหมายการรักษาแตกต่างกันไปตามชนิด ในชนิดสมบูรณ์และไม่สมบูรณ์ เป้าหมายหลักคือการสร้างเสริมความสวยงาม และการพยากรณ์โรคในการฟื้นฟูการมองเห็น มีจำกัดมาก ในชนิดไม่สมบูรณ์ การจัดการความเสี่ยงของโรคกระจกตา จากการสัมผัสและความบกพร่องทางการมองเห็น เป็นเรื่องเร่งด่วน
การจัดการผิวตา : จ่ายสารหล่อลื่นตาและน้ำตาเทียม (เพื่อป้องกันการสัมผัสและความแห้ง) หากมีความบกพร่องขนาดใหญ่ ให้ใช้ยาขี้ผึ้งทาตาเพื่อป้องกันกระจกตา แห้งเปลือกตาเทียม : ทางเลือกเมื่อการผ่าตัดมีข้อห้าม ไม่สามารถทำได้ หรือล้มเหลว
ชนิดสมบูรณ์
ระยะที่ 1 : หลังจากกรีดผิวหนังส่วนที่เหลือของลูกตา ใส่คอนฟอร์เมอร์ ที่หุ้มด้วยแผ่นเยื่อเมือกเพื่อสร้างถุงเยื่อบุตา
ระยะที่ 2 (ประมาณ 1 ปีต่อมา): ทำการสร้างหนังตาขึ้นใหม่ พร้อมเสริมแผ่นชั้นหลังและปลูกถ่ายเยื่อเมือกเบ้าตา หากการปลูกถ่ายเยื่อเมือกไม่ได้ผล อาจใช้หนังหุ้มปลายอวัยวะเพศเป็นทางเลือก
ชนิดไม่สมบูรณ์และบกพร่อง
ชนิดไม่สมบูรณ์ : หลังจากสร้างถุงเยื่อบุตา (แผ่นเยื่อเมือก + วางคอนฟอร์เมอร์ ) จะทำการสร้างหนังตาขึ้นใหม่ โดยใช้การผ่าตัดแบ่งหนังตาหรือวิธีสลับ มีความเสี่ยงที่หนังตาจะยึดติดกับกระจกตา ซ้ำ
ชนิดไม่สมบูรณ์ : เป้าหมายหลักคือการสร้างหนังตาบนและฟอร์นิกซ์บนขึ้นใหม่ ทำการผ่าตัดสร้างใหม่ในครั้งเดียวโดยใช้การปลูกถ่ายตาขาว หรือเยื่อหุ้มน้ำคร่ำ
หากความบกพร่องในเด็กมีขนาดเล็ก สามารถซ่อมแซมด้วยการเย็บแบบต่อกันได้ หากมีขนาดใหญ่ จะเลือกการผ่าตัดตกแต่งด้วยแผ่นผิวหนัง ในรายงานผู้ป่วยเด็ก รายหนึ่ง พบลูกตาชนิดถุงน้ำพื้นฐานที่ติดอยู่ระหว่างการสำรวจ และไม่มีการผ่าตัดเพิ่มเติม1)
ในชนิดสมบูรณ์และไม่สมบูรณ์ การมองเห็น ดีขึ้นนั้นพบได้น้อยมาก จำเป็นต้องอธิบายให้ครอบครัวผู้ป่วยเข้าใจอย่างเพียงพอ
การผ่าตัดทำเป็นขั้นตอน โดยให้แน่ใจว่าแต่ละขั้นตอนหายดีแล้วจึงดำเนินการขั้นต่อไป
หลังผ่าตัด ความเสี่ยงของการกลับเป็นซ้ำของการยึดติดระหว่างกระจกตา และหนังตายังคงมีอยู่ การติดตามผลอย่างสม่ำเสมอเป็นสิ่งจำเป็น
Q
การผ่าตัดตาแบบซ่อนสมบูรณ์ทำอย่างไร?
A
ทำเป็นขั้นตอน ขั้นแรก หลังจากกรีดผิวหนังเหนือเศษลูกตา ใส่คอนฟอร์เมอร์ ที่หุ้มด้วยเยื่อบุปลูกถ่ายเพื่อสร้างถุงเยื่อบุตา ประมาณหนึ่งปีต่อมา ในขั้นที่สอง ทำการสร้างเปลือกตาขึ้นใหม่พร้อมเสริมแผ่นชั้นหลังและปลูกถ่ายเยื่อบุเบ้าตา เป้าหมายหลักคือการปรับปรุงรูปลักษณ์ด้านความสวยงาม และการพยากรณ์โรคในการมองเห็น ดีขึ้นนั้นพบได้น้อยมาก
ยังไม่มีฉันทามติที่ชัดเจนเกี่ยวกับพยาธิสรีรวิทยา
ความบกพร่องของนิวโรเอ็กโทเดิร์ม : ถุงตาจากนิวโรเอ็กโทเดิร์มจำเป็นต่อการพัฒนาเลนส์ของทารกในครรภ์ และความบกพร่องในชั้นนี้ขัดขวางการพัฒนาที่เหมาะสมของกระจกตา เลนส์ และช่องหน้าม่านตา ความผิดปกติของการสร้างเปลือกตา : เปลือกตาจะไม่เกิดขึ้นหากไม่มีการแบ่งตัวของเอ็กโทเดิร์มและเมโซเดิร์ม เปลือกตาปรากฏในสัปดาห์ที่ 6 ของการตั้งครรภ์ และเปลือกตาบนและล่างจะเชื่อมกันจนถึงเดือนที่ 7 ความล้มเหลวในการสร้างในช่วงนี้ทำให้เกิดความผิดปกติของเปลือกตาแต่กำเนิดทฤษฎีข้อบกพร่องของอะพอพโทซิส : การพบร่วมกับนิ้วติดกัน ความผิดปกติของกล่องเสียงและอวัยวะสืบพันธุ์ ชี้ให้เห็นว่าข้อบกพร่องในการตายของเซลล์แบบโปรแกรม มีบทบาทสำคัญความผิดปกติของสารเชิงซ้อน FRAS/FREM : การยึดเกาะที่บกพร่องระหว่างเยื่อฐานและเยื่อบุผิวเนื่องจากการกลายพันธุ์ของ FRAS1, FREM1 และ FREM2 ทำให้เปลือกตาแยกไม่สำเร็จ1)
ภาวะแทรกซ้อนทุติยภูมิที่พบบ่อยร่วมกับตาเล็ก ได้แก่ ต้อกระจก เลนส์เคลื่อน ต้อหิน และจอประสาทตาลอก
เนื้อหาต่อไปนี้อยู่ในขั้นตอนการวิจัยหรือการทดลองทางคลินิกในปัจจุบัน และไม่ใช่การรักษามาตรฐานที่สามารถรับได้ในโรงพยาบาลทั่วไป เป็นข้อมูลอ้างอิงสำหรับผู้เชี่ยวชาญเกี่ยวกับการพัฒนาทางการแพทย์ในอนาคต
Mwipopo และคณะ (2023) รายงานกรณีตาซ่อนสองข้างจากแทนซาเนีย 1) การตรวจทางพันธุกรรมระบุการกลายพันธุ์ FREM2 (น่าจะก่อโรคแบบเฮเทอโรไซกัส) ร่วมกับการกลายพันธุ์ CEP85L (สมองเรียบ 10: LIS10) LIS10 แสดงความหลากหลายตั้งแต่ภาวะปัญญาอ่อนเล็กน้อยไปจนถึงฟีโนไทป์รุนแรง กรณีนี้เป็นรายงานแรกจากแอฟริกา และมีการชี้ให้เห็นว่าการขาดรายงานในประเทศที่มีรายได้ต่ำและปานกลางมีส่วนทำให้จำนวนผู้ป่วยต่ำกว่าความเป็นจริง
เนื่องจากการกลายพันธุ์ของยีนที่เกี่ยวข้องกับคอมเพล็กซ์ FRAS/FREM แสดงฟีโนไทป์ที่ทับซ้อนกันในหลายกลุ่มอาการ การวินิจฉัยทางคลินิกเพียงอย่างเดียวไม่เพียงพอ และบทบาทของการตรวจทางพันธุกรรมเพิ่มขึ้น 1) ความจำเป็นในการตรวจทางพันธุกรรมก็เพิ่มขึ้นในประเทศที่มีรายได้ต่ำและปานกลางเช่นกัน 1)
Mwipopo E, Massomo MM, Moshiro R, Manji KP. Bilateral cryptophthalmos with overlapping features of Manitoba oculo-tricho-anal (MOTA) syndrome and Fraser syndrome 2. BMJ Case Rep. 2023;16(7):e252618.
Thomas IT, Frias JL, Felix V, Sanchez de Leon L, Hernandez RA, Jones MC. Isolated and syndromic cryptophthalmos. Am J Med Genet. 1986;25(1):85-98. PMID: 3099574.
Kabra M, Gulati S, Ghosh M, Menon PS. Fraser-cryptophthalmos syndrome. Indian J Pediatr. 2000;67(10):775-8. PMID: 11105430.
ถาม AI เกี่ยวกับบทความนี้
คัดลอกข้อความบทความแล้ววางในผู้ช่วย AI ที่คุณต้องการใช้
เปิดผู้ช่วย AI ด้านล่าง แล้ววางข้อความที่คัดลอกลงในช่องแชต