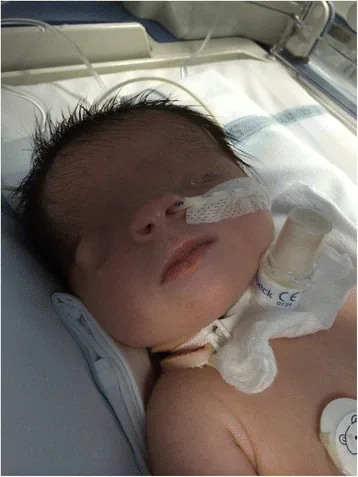
Cryptophthalmos trong tiếng Latin có nghĩa là “mắt ẩn”. Đây là tình trạng da mi mắt dính liền che phủ nhãn cầu và hốc mắt, không có khe mi. Da trán liên tục với da má.
Tỷ lệ mắc được báo cáo là 0,043 trên 10.000 trẻ sinh sống và 1,1 trên 10.000 trẻ chết lưu1). Tính đến năm 2018, chỉ có 55 ca được báo cáo trong y văn. Tỷ lệ hiện mắc chung của các bất thường mi mắt là 0,06%, với 2/3 trường hợp lẻ tẻ và 1/3 có yếu tố di truyền1).
Cryptophthalmos được phân loại thành ba thể dựa trên hình thái.
Thể hoàn toàn (điển hình): Tắc hoàn toàn hốc mắt. Thể nặng nhất với không có lông mày, lông mi và cấu trúc tuyến.
Thể không hoàn toàn (không điển hình): Còn mi mắt dấu vết. Có túi kết mạc nhỏ ở phía ngoài.
Thể thoái triển (dạng abortive / dính mi cầu bẩm sinh): Thiếu mi trên. Da trán dính vào phần trên giác mạc.
Bất kỳ thể nào cũng có thể một bên hoặc hai bên, đơn độc hoặc trong hội chứng. Mắt ẩn là mắt bị che phủ bởi da mi dính, có thể xuất hiện đơn độc hoặc là một trong các triệu chứng của hội chứng Fraser. Nếu bờ mi không hình thành, được chẩn đoán là vô mi hoặc mắt ẩn.
Liên quan rất chặt chẽ với hội chứng Fraser, phần lớn là di truyền lặn nhiễm sắc thể thường do đột biến gen FRAS1, GRIP1 và FREM2. Mắt ẩn (cryptophthalmos) được tìm thấy ở 80-93% các trường hợp hội chứng Fraser.
QTrẻ bị mắt ẩn có thể phục hồi thị lực không?
A
Hầu như tất cả các trường hợp đều không có hoặc rất ít thị lực. Mục đích của phẫu thuật chủ yếu là cải thiện thẩm mỹ và tái tạo vùng mắt, rất hiếm khi cải thiện được thị lực. Xem phần “Phương pháp điều trị tiêu chuẩn” để biết chi tiết.
De Bernardo G, Giordano M, Di Toro A, et al. Prenatal diagnosis of Fraser syndrome: a matter of life or death? Ital J Pediatr. 2015 Nov 9;41:86. Figure 1. PMCID: PMC4640198. License: CC BY.
Ảnh chụp khuôn mặt ngay sau sinh, khe mi mắt không hình thành và da che phủ bề mặt nhãn cầu. Mắt trái là mắt nhỏ, mắt phải cũng có bất thường về hình thái, giúp hiểu được ngoại hình của mắt ẩn.
Dưới đây là các dấu hiệu lâm sàng chính theo từng loại.
Thể hoàn toàn
Tắc hốc mắt: Da trán và má liên tục, khe mi mắt hoàn toàn không có.
Thiếu phần phụ: Lông mày, lông mi và cấu trúc tuyến không tồn tại.
Kết hợp nhãn cầu nhỏ: Da bề mặt dính liền với giác mạc. Không có túi kết mạc, nhãn cầu nhỏ rất phổ biến. Có thể có nang hốc mắt.
Thể không hoàn toàn và thể thiếu
Thể không hoàn toàn: Mi mắt dạng vết tích còn lại. Chiều dài khe mi khoảng 1/3 bình thường. Có một túi kết mạc nhỏ ở phía ngoài. Nhãn cầu nhỏ và gần như bị da che phủ.
Thể không hoàn chỉnh: Không có mí mắt trên. Da trán dính vào 75% phần trên của giác mạc. Giác mạc bị che phủ bị sừng hóa và đục, nhưng giác mạc lộ ra có thể trong suốt.
Trong một báo cáo ca bệnh, một bé trai 39 tuần tuổi thai có biểu hiện mắt ẩn hai bên, không có lông mày, đường chân tóc trước bất thường, khe hở đầu mũi, tai thấp, mắt cách xa nhau và dị dạng hậu môn trực tràng thấp 1). Sờ thấy một nang hốc mắt di động kích thước 1×1 cm ở mắt phải, và mắt trái bị chìm sâu 1). CT cho thấy mô hình hồi não bất thường ở vùng chẩm và mắt trái nhỏ hơn 1).
Nguyên nhân của mắt ẩn là đột biến gen ở các protein cấu thành phức hợp FRAS/FREM.
Phức hợp FRAS/FREM: Các protein FRAS1, FREM1 và FREM2 duy trì sự kết dính giữa màng đáy và biểu mô trong quá trình phát triển phôi. Đột biến gây rối loạn kết dính, dẫn đến không tách rời mí mắt 1).
Hội chứng Fraser (FRASRS1/2): Di truyền lặn nhiễm sắc thể thường do đột biến gen FRAS1 hoặc FREM2 1).
Hội chứng MOTA: do đột biến gen FREM1 gây ra, bao gồm khe hở mũi, dị tật hậu môn-trực tràng, bất sản thận, v.v. 1).
Mắt ẩn đơn độc: Đột biến FREM2 cũng có thể gây ra mắt ẩn đơn độc một bên hoặc hai bên1).
Kiểu di truyền: Thường là di truyền lặn trên nhiễm sắc thể thường. Cũng có báo cáo về di truyền trội trên nhiễm sắc thể thường.
Các cơ chế được biết đến gây ra khuyết tật bẩm sinh mí mắt bao gồm sự đóng không hoàn toàn của các khe hở mặt trong thời kỳ phôi thai và sự chèn ép từ dải ối.
QCryptophthalmos liên quan đến hội chứng Fraser như thế nào?
A
Cryptophthalmos được tìm thấy trong 80-93% trường hợp hội chứng Fraser, và đột biến gen FRAS1/FREM2 là nguyên nhân chính. Phức hợp FRAS/FREM rất cần thiết để duy trì sự kết dính giữa màng đáy và biểu mô trong quá trình phát triển phôi, và đột biến của nó dẫn đến sự thất bại trong việc tách mí mắt 1). Tuy nhiên, đột biến FREM2 cũng có thể gây ra cryptophthalmos đơn độc.
Có thể phát hiện bằng siêu âm trước sinh vào khoảng tuần thứ 18 của thai kỳ. Các dấu hiệu bao gồm không có khe mí mắt và da liên tục từ trán đến má. Nếu có dính ngón, tăng âm phổi và thiểu ối, khả năng cao là hội chứng Fraser.
Xét nghiệm bảng gen FRAS/FREM rất quan trọng để xác định chẩn đoán1). Vì kiểu hình của hội chứng Fraser và hội chứng MOTA chồng lấn, nên khó phân biệt chỉ dựa trên các dấu hiệu lâm sàng mà không có xét nghiệm di truyền.
Chẩn đoán phân biệt bao gồm khuyết tật bẩm sinh mí mắt (trong trường hợp khuyết một phần), một mắt, và bất đẳng nhãn cầu.
Mục tiêu điều trị khác nhau tùy theo loại. Ở loại hoàn toàn và không hoàn toàn, mục tiêu chính là tái tạo thẩm mỹ, và tiên lượng cải thiện thị lực rất hạn chế. Ở loại không hoàn chỉnh, việc xử lý nguy cơ bệnh lý giác mạc do lộ và suy giảm thị lực là cấp bách.
Quản lý bề mặt nhãn cầu: Kê đơn chất bôi trơn mắt và nước mắt nhân tạo (để chống lộ và khô). Nếu khuyết tật lớn, sử dụng thuốc mỡ tra mắt để ngăn khô giác mạc.
Mắt giả che: Lựa chọn khi phẫu thuật chống chỉ định, không thể thực hiện hoặc thất bại.
Giai đoạn 1: Sau khi rạch da phần còn lại của nhãn cầu, đặt một dụng cụ định hình được phủ bằng mảnh ghép niêm mạc để tạo túi kết mạc.
Giai đoạn 2 (khoảng 1 năm sau): Tiến hành tái tạo mi mắt với tăng cường lớp sau và ghép niêm mạc hốc mắt. Nếu ghép niêm mạc không hiệu quả, có thể sử dụng bao quy đầu làm vật thay thế.
Loại không hoàn toàn và thiếu
Loại không hoàn toàn: Sau khi tạo túi kết mạc (ghép niêm mạc + đặt dụng cụ định hình), tái tạo mi mắt được thực hiện bằng phẫu thuật chia sẻ mi hoặc phương pháp chuyển đổi. Có nguy cơ tái phát dính mi mắt-giác mạc.
Loại không hoàn toàn: Mục tiêu chính là tái tạo mi trên và vòm trên. Phẫu thuật tái tạo một thì được thực hiện bằng cách ghép củng mạc hoặc màng ối.
Nếu khuyết tật nhỏ ở trẻ em, có thể sửa chữa bằng khâu nối trực tiếp. Nếu lớn, phẫu thuật tạo hình bằng vạt da được lựa chọn. Trong trường hợp báo cáo, một nhãn cầu dạng nang thô sơ dính đã được phát hiện khi thăm dò, và không có phẫu thuật bổ sung nào được thực hiện1).
QPhẫu thuật cho mắt ẩn hoàn toàn được thực hiện như thế nào?
A
Được thực hiện theo từng giai đoạn. Giai đoạn đầu, sau khi rạch da trên phần còn lại của nhãn cầu, một dụng cụ định hình được bọc bằng mảnh ghép niêm mạc được đưa vào để tạo túi kết mạc. Khoảng một năm sau, ở giai đoạn thứ hai, tiến hành tái tạo mí mắt với tăng cường lớp sau và ghép niêm mạc hốc mắt. Mục tiêu chính là cải thiện ngoại hình thẩm mỹ, và tiên lượng cải thiện thị lực là cực kỳ hiếm.
Khiếm khuyết ngoại bì thần kinh: Túi thị giác ngoại bì thần kinh rất cần thiết cho sự phát triển của thủy tinh thể thai nhi, và khiếm khuyết ở lớp này ngăn cản sự phát triển thích hợp của giác mạc, thủy tinh thể và tiền phòng.
Rối loạn hình thành mí mắt: Mí mắt không hình thành nếu không có sự biệt hóa của ngoại bì và trung bì. Mí mắt xuất hiện vào tuần thứ 6 của thai kỳ, và mí trên và mí dưới dính liền cho đến tháng thứ 7. Sự thất bại trong hình thành trong giai đoạn này gây ra các bất thường mí mắt bẩm sinh.
Thuyết khiếm khuyết apoptosis: Sự kết hợp của dính ngón tay, bất thường thanh quản và cơ quan sinh dục cho thấy khiếm khuyết trong quá trình chết tế bào theo chương trình đóng vai trò quan trọng.
Rối loạn chức năng phức hợp FRAS/FREM: Sự bám dính bị suy yếu giữa màng đáy và biểu mô do đột biến FRAS1, FREM1 và FREM2 dẫn đến không tách rời được mí mắt1).
Các biến chứng thứ phát thường gặp kèm theo mắt nhỏ bao gồm đục thủy tinh thể, lệch thủy tinh thể, glôcôm và bong võng mạc.
7. Nghiên cứu mới nhất và triển vọng tương lai (báo cáo giai đoạn nghiên cứu)
Mwipopo và cộng sự (2023) đã báo cáo một trường hợp mắt ẩn hai bên từ Tanzania 1). Xét nghiệm di truyền xác định đột biến FREM2 (có khả năng gây bệnh dị hợp tử) cùng với đột biến CEP85L (não nhẵn 10: LIS10). LIS10 cho thấy sự không đồng nhất từ chậm phát triển tâm thần nhẹ đến kiểu hình nặng. Trường hợp này là báo cáo đầu tiên từ châu Phi, và được chỉ ra rằng việc thiếu báo cáo ở các nước thu nhập thấp và trung bình góp phần làm giảm ước tính số ca.
Vì các đột biến gen liên quan đến phức hợp FRAS/FREM cho thấy kiểu hình chồng chéo trong nhiều hội chứng, chẩn đoán lâm sàng đơn thuần là không đủ, và vai trò của xét nghiệm di truyền ngày càng tăng 1). Nhu cầu xét nghiệm di truyền cũng tăng lên ở các nước thu nhập thấp và trung bình 1).
Mwipopo E, Massomo MM, Moshiro R, Manji KP. Bilateral cryptophthalmos with overlapping features of Manitoba oculo-tricho-anal (MOTA) syndrome and Fraser syndrome 2. BMJ Case Rep. 2023;16(7):e252618.
Thomas IT, Frias JL, Felix V, Sanchez de Leon L, Hernandez RA, Jones MC. Isolated and syndromic cryptophthalmos. Am J Med Genet. 1986;25(1):85-98. PMID: 3099574.
Kabra M, Gulati S, Ghosh M, Menon PS. Fraser-cryptophthalmos syndrome. Indian J Pediatr. 2000;67(10):775-8. PMID: 11105430.
Sao chép toàn bộ bài viết và dán vào trợ lý AI bạn muốn dùng.
Đã sao chép bài viết vào clipboard
Mở một trợ lý AI bên dưới và dán nội dung đã sao chép vào ô chat.