Criptoftalmia significa “olho oculto” em latim. Refere-se a uma condição em que a pele palpebral fundida cobre o globo ocular e a órbita, com ausência da fissura palpebral. A pele da testa é contínua com a pele da bochecha.
A incidência relatada é de 0,043 por 10.000 nascidos vivos e 1,1 por 10.000 natimortos1). Até 2018, apenas 55 casos foram relatados na literatura. A prevalência geral de anomalias palpebrais é de 0,06%, com 2/3 dos casos esporádicos e 1/3 com fator genético1).
O criptoftalmo é classificado em três tipos de acordo com a morfologia.
Tipo completo (típico): Oclusão total da órbita. Tipo mais grave com ausência de sobrancelhas, cílios e estruturas glandulares.
Tipo incompleto (atípico): Pálpebras vestigiais remanescentes. Pequeno saco conjuntival no lado lateral.
Tipo abortivo (forma abortiva / sinéquia palpebro-ocular congênita): Ausência da pálpebra superior. Pele da testa aderida à parte superior da córnea.
Qualquer tipo pode ser unilateral ou bilateral, isolado ou sindrômico. Olho oculto é o olho coberto por pele palpebral fundida, podendo aparecer isoladamente ou como um dos sintomas da síndrome de Fraser. Se a margem palpebral não estiver formada, diagnostica-se como anoftalmia ou olho oculto.
Está intimamente associado à síndrome de Fraser, sendo na maioria das vezes uma herança autossômica recessiva devido a mutações nos genes FRAS1, GRIP1 e FREM2. O criptoftalmo é observado em 80-93% dos casos da síndrome de Fraser.
QA visão de uma criança com criptoftalmo pode ser recuperada?
A
Em quase todos os casos, não há visão ou é muito limitada. O objetivo da cirurgia é principalmente a melhora estética e a reconstrução da região ocular, sendo extremamente raro obter melhora visual. Consulte a seção “Métodos de Tratamento Padrão” para detalhes.
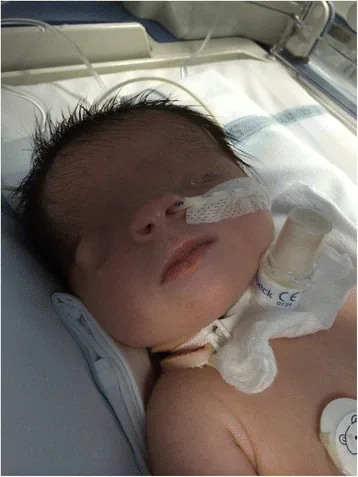
De Bernardo G, Giordano M, Di Toro A, et al. Prenatal diagnosis of Fraser syndrome: a matter of life or death? Ital J Pediatr. 2015 Nov 9;41:86. Figure 1. PMCID: PMC4640198. License: CC BY.
Fotografia facial logo após o nascimento, mostrando que a fenda palpebral não se formou e a pele cobre a superfície ocular. O olho esquerdo é microftálmico, e o olho direito também apresenta anomalias morfológicas, ajudando a entender a aparência do criptoftalmo.
Abaixo estão os principais achados clínicos por tipo.
Tipo completo
Oclusão orbitária: A pele da testa e da bochecha são contínuas, com ausência completa da fissura palpebral.
Ausência de anexos: Sobrancelhas, cílios e estruturas glandulares estão ausentes.
Associação com microftalmia: A pele superficial funde-se com a córnea. Não há saco conjuntival, e a microftalmia é muito comum. Podem existir cistos orbitários.
Tipo incompleto e tipo parcial
Tipo incompleto: Pálpebras vestigiais permanecem. O comprimento da fissura palpebral é cerca de 1/3 do normal. Há um pequeno saco conjuntival lateralmente. O globo ocular é pequeno e quase totalmente coberto pela pele.
Tipo incompleto: Ausência da pálpebra superior. Pele da testa aderida a 75% da parte superior da córnea. A córnea coberta torna-se queratinizada e opaca, mas a córnea exposta pode ser transparente.
Em um relato de caso, um menino com 39 semanas de gestação apresentou criptoftalmia bilateral, ausência de sobrancelhas, linha capilar frontal irregular, fenda na ponta do nariz, orelhas baixas, hipertelorismo e malformação anorretal baixa 1). Um cisto orbitário móvel de 1×1 cm foi palpado no olho direito, e o olho esquerdo estava profundamente enterrado 1). A TC mostrou padrão anormal de giros occipitais e olho esquerdo reduzido 1).
A causa da criptoftalmia são mutações genéticas nas proteínas que compõem o complexo FRAS/FREM.
Complexo FRAS/FREM: As proteínas FRAS1, FREM1 e FREM2 mantêm a adesão entre a membrana basal e o epitélio durante o desenvolvimento embrionário. Mutações causam falha de adesão, resultando em não separação das pálpebras 1).
Síndrome de Fraser (FRASRS1/2): Herança autossômica recessiva devido a mutações nos genes FRAS1 ou FREM2 1).
Síndrome MOTA: causada por mutação no FREM1, associada a fenda nasal, malformações anorretais, agenesia renal, entre outros 1).
Olho oculto isolado: A mutação FREM2 também pode causar olho oculto isolado unilateral ou bilateral1).
Padrão de herança: Frequentemente autossômico recessivo. Também há relatos de herança autossômica dominante.
Os mecanismos conhecidos para a ocorrência de coloboma palpebral congênito incluem o fechamento incompleto das fissuras faciais durante o período embrionário e a compressão por bandas amnióticas.
QComo o criptoftalmo está relacionado à síndrome de Fraser?
A
O criptoftalmo está presente em 80-93% dos casos de síndrome de Fraser, sendo as mutações nos genes FRAS1/FREM2 a principal causa. O complexo FRAS/FREM é essencial para manter a adesão entre a membrana basal e o epitélio durante o desenvolvimento embrionário, e suas mutações levam à falha na separação das pálpebras 1). No entanto, mutações no FREM2 também podem causar criptoftalmo isolado.
Pode ser detectado por ultrassonografia pré-natal por volta da 18ª semana de gestação. Os achados incluem ausência da fissura palpebral e pele contínua da testa à bochecha. Se houver sindactilia, aumento da ecogenicidade pulmonar e oligoidrâmnio, a probabilidade de síndrome de Fraser é alta.
Os critérios diagnósticos para a síndrome de Fraser são apresentados abaixo.
Classificação
Item
Critério principal
Criptoftalmia, sindactilia, anomalias genitais, anomalias de membros
Critério secundário
Anomalias de orelha e nariz, lábio leporino e fenda palatina, anomalia da linha do cabelo, anomalias renais
Exames necessários:
Exame de acuidade visual, estrabismo e fundo de olho: Avaliação da função visual
Teste de tração sob anestesia geral: Confirmação de bandas fibrosas ocultas
TC/RM: Avaliação da morfologia do globo ocular e do cérebro. Útil para confirmar complicações do sistema nervoso central, como padrão anormal de giros occipitais1)
O painel FRAS/FREM é importante para confirmar o diagnóstico1). Como os fenótipos da síndrome de Fraser e da síndrome de MOTA se sobrepõem, pode ser difícil diferenciá-los apenas com base nos achados clínicos sem o teste genético.
O diagnóstico diferencial inclui coloboma palpebral congênito (no caso de defeito parcial), ciclopia e anisofthalmia.
O objetivo do tratamento varia conforme o tipo. Nos tipos completo e incompleto, o objetivo principal é a reconstrução estética, e o prognóstico para melhora visual é muito limitado. No tipo abortivo, o manejo do risco de ceratopatia de exposição e deficiência visual é urgente.
Manejo da superfície ocular: Prescrição de lubrificantes oculares e lágrimas artificiais (para exposição e ressecamento). Em caso de defeito grande, use pomada oftálmica para prevenir ressecamento da córnea.
Prótese palpebral: Opção quando a cirurgia é contraindicada, impossível ou malsucedida.
1ª Etapa: Após incisão cutânea do remanescente ocular, insere-se um conformador coberto por enxerto de mucosa para criar o saco conjuntival.
2ª Etapa (cerca de 1 ano depois): Realiza-se reconstrução palpebral com reforço da lâmina posterior e transplante de mucosa orbitária. Se o enxerto de mucosa falhar, o prepúcio pode ser usado como alternativa.
Tipo Incompleto e Deficiente
Tipo Incompleto: Após a criação do saco conjuntival (enxerto de mucosa + colocação de conformador), a reconstrução palpebral é feita por cirurgia de compartilhamento palpebral ou método de switch. Há risco de recorrência de aderência córneo-palpebral.
Tipo incompleto: O objetivo principal é a reconstrução da pálpebra superior e do fórnice superior. A reconstrução em um estágio é realizada com transplante de esclera ou âmnio.
Se o defeito for pequeno em uma criança, pode ser reparado com sutura término-terminal. Se for grande, a cirurgia plástica com retalho cutâneo é escolhida. No caso relatado, um olho cístico rudimentar aderido foi encontrado durante a exploração, e nenhuma cirurgia adicional foi realizada1).
QComo é realizada a cirurgia para olho oculto completo?
A
É realizada em etapas. Na primeira etapa, após incisão da pele sobre o resíduo ocular, um conformador revestido com enxerto de mucosa é inserido para criar o saco conjuntival. Cerca de um ano depois, na segunda etapa, é realizada a reconstrução palpebral com reforço da lâmina posterior e enxerto de mucosa orbitária. O objetivo principal é a melhora estética, e o prognóstico de melhora visual é extremamente raro.
Ainda não há consenso estabelecido sobre a fisiopatologia.
Defeito neuroectodérmico: A vesícula óptica neuroectodérmica é essencial para o desenvolvimento do cristalino fetal, e um defeito nessa camada impede o desenvolvimento adequado da córnea, cristalino e câmara anterior.
Distúrbio da formação palpebral: A pálpebra não se forma sem a diferenciação do ectoderma e mesoderma. A pálpebra surge na 6ª semana de gestação, e as pálpebras superior e inferior permanecem fundidas até o 7º mês. A falha na formação durante esse período causa anomalias palpebrais congênitas.
Teoria do defeito de apoptose: A associação com sindactilia, anomalias laríngeas e genitais sugere que um defeito na morte celular programada desempenha um papel importante.
Disfunção do complexo FRAS/FREM: A adesão prejudicada entre a membrana basal e o epitélio devido a mutações em FRAS1, FREM1 e FREM2 leva à falha na separação palpebral1).
Mwipopo et al. (2023) relataram um caso de olho oculto bilateral da Tanzânia 1). O teste genético identificou uma mutação FREM2 (provavelmente patogênica heterozigótica) juntamente com uma mutação CEP85L (lisencefalia 10: LIS10). A LIS10 mostra heterogeneidade, desde retardo mental leve até fenótipos graves. Este caso é o primeiro relato da África, e foi apontado que a falta de notificação em países de baixa e média renda contribui para a subestimação do número de casos.
Como as mutações genéticas relacionadas ao complexo FRAS/FREM mostram fenótipos sobrepostos em múltiplas síndromes, o diagnóstico clínico isolado é insuficiente, e o papel do teste genético está aumentando 1). A necessidade de teste genético também está aumentando em países de baixa e média renda 1).
Mwipopo E, Massomo MM, Moshiro R, Manji KP. Bilateral cryptophthalmos with overlapping features of Manitoba oculo-tricho-anal (MOTA) syndrome and Fraser syndrome 2. BMJ Case Rep. 2023;16(7):e252618.
Thomas IT, Frias JL, Felix V, Sanchez de Leon L, Hernandez RA, Jones MC. Isolated and syndromic cryptophthalmos. Am J Med Genet. 1986;25(1):85-98. PMID: 3099574.
Kabra M, Gulati S, Ghosh M, Menon PS. Fraser-cryptophthalmos syndrome. Indian J Pediatr. 2000;67(10):775-8. PMID: 11105430.
Copie o texto do artigo e cole no assistente de IA de sua preferência.
Artigo copiado para a área de transferência
Abra um assistente de IA abaixo e cole o texto copiado na conversa.