Kryptophthalmus bedeutet aus dem Lateinischen übersetzt „verborgenes Auge“. Es bezeichnet einen Zustand, bei dem die verwachsene Augenlidhaut den Augapfel und die Augenhöhle bedeckt und die Lidspalte fehlt. Die Stirnhaut geht kontinuierlich in die Wangenhaut über.
Die Inzidenz wird mit 0,043 pro 10.000 Lebendgeburten und 1,1 pro 10.000 Totgeburten angegeben1). Bis 2018 wurden nur 55 Fälle in der Literatur beschrieben. Die Prävalenz aller Lidfehlbildungen beträgt 0,06 %, wobei zwei Drittel sporadisch und ein Drittel genetisch bedingt sind1).
Kryptophthalmus wird je nach Morphologie in drei Typen eingeteilt.
Vollständiger Typ (typisch): Vollständiger Verschluss der Augenhöhle. Schwerste Form mit Fehlen von Augenbrauen, Wimpern und Drüsenstrukturen.
Unvollständiger Typ (atypisch): Restliche Augenlider sind vorhanden. Lateral besteht ein kleiner Bindehautsack.
Unvollständige Form (abortive Form / angeborene Lid-Kornea-Verwachsung): Fehlen des Oberlids. Die Stirnhaut haftet am oberen Teil der Hornhaut.
Beide Typen können einseitig oder beidseitig, isoliert oder syndromal auftreten. Beim Kryptophthalmus ist der Augapfel von verwachsener Lidhaut bedeckt; er kann isoliert oder als eines der Symptome des Fraser-Syndroms auftreten. Wenn der Lidrand vollständig fehlt, wird die Diagnose Ankyloblepharon oder Kryptophthalmus gestellt.
Die Assoziation mit dem Fraser-Syndrom ist sehr eng; häufig liegt eine autosomal-rezessive Vererbung mit Mutationen in den Genen FRAS1, GRIP1 und FREM2 vor. Bei 80–93 % der Patienten mit Fraser-Syndrom findet sich ein Kryptophthalmus.
QKann ein Kind mit Kryptophthalmus das Sehvermögen wiedererlangen?
A
In fast allen Fällen ist das Sehvermögen kaum oder gar nicht vorhanden. Ziel der Operation ist hauptsächlich die kosmetische Verbesserung des Erscheinungsbildes und die Rekonstruktion der Augenregion; eine Verbesserung des Sehvermögens ist äußerst selten. Einzelheiten finden Sie im Abschnitt „Standardbehandlungen“.
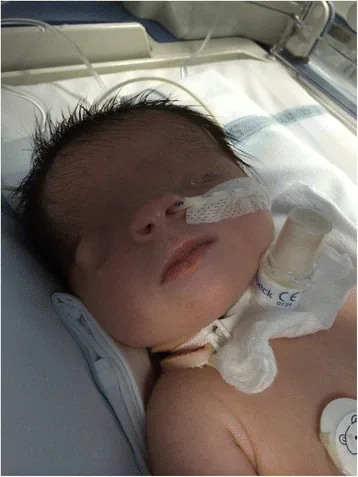
De Bernardo G, Giordano M, Di Toro A, et al. Prenatal diagnosis of Fraser syndrome: a matter of life or death? Ital J Pediatr. 2015 Nov 9;41:86. Figure 1. PMCID: PMC4640198. License: CC BY.
Gesichtsaufnahme direkt nach der Geburt: Die Lidspalte ist nicht ausgebildet, und die Haut bedeckt die Augenoberfläche. Links liegt ein Mikrophthalmus vor, auch das rechte Auge zeigt Formanomalien, was das Verständnis des Erscheinungsbildes des Kryptophthalmus erleichtert.
Die wichtigsten klinischen Befunde nach Typ sind im Folgenden aufgeführt.
Vollständiger Typ
Orbitalverschluss: Die Haut von Stirn und Wange ist durchgehend, die Lidspalte fehlt vollständig.
Fehlen der Augenanhangsgebilde: Augenbrauen, Wimpern und Drüsenstrukturen fehlen.
Assoziierte Mikrophthalmie: Die Hautoberfläche ist mit der Hornhaut verwachsen. Es gibt keinen Bindehautsack, Mikrophthalmie ist sehr häufig. Orbitazysten können vorhanden sein.
Unvollständige Form
Unvollständige Form: Reste der Augenlider sind vorhanden. Die Lidspaltenlänge beträgt etwa ein Drittel der normalen. Lateral gibt es einen kleinen Bindehautsack. Der Augapfel ist klein und fast vollständig von Haut bedeckt.
Inkomplette Form: Fehlen des Oberlids. Die Stirnhaut haftet an den oberen 75% der Hornhaut. Die bedeckte Hornhaut kann keratinisieren und eintrüben, die exponierte Hornhaut kann jedoch klar sein.
In einem Fallbericht wurde bei einem männlichen Neugeborenen in der 39. Schwangerschaftswoche beidseitiges Kryptophthalmus, Fehlen der Augenbrauen, unregelmäßige Stirnhaarlinie, Nasenspitzenriss, tief sitzende Ohrmuscheln, Hypertelorismus und tief sitzende anorektale Fehlbildung festgestellt1). Am rechten Auge war eine 1×1 cm große bewegliche Orbitazyste tastbar, der linke Augapfel war tief eingebettet1). Im CT zeigten sich ein abnormes Gyrierungsmuster im Hinterkopf und eine Verkleinerung des linken Augapfels1).
Die Ursache des Kryptophthalmus sind Genmutationen in Proteinen, die den FRAS/FREM-Komplex bilden.
FRAS/FREM-Komplex: Die Proteine FRAS1, FREM1 und FREM2 erhalten während der Embryonalentwicklung die Adhäsion zwischen Basalmembran und Epithel. Mutationen führen zu Adhäsionsstörungen und verursachen eine Ankyloblepharon1).
Fraser-Syndrom (FRASRS1/2): Autosomal-rezessive Vererbung durch Mutationen in FRAS1 oder FREM21).
MOTA-Syndrom: FREM1-Mutationen führen zu Nasenspalte, anorektalen Fehlbildungen, Nierenagenesie u.a.1).
Isoliertes Kryptophthalmus: FREM2-Mutationen können auch Ursache für einseitigen oder beidseitigen isolierten Kryptophthalmus sein1).
Vererbungsmuster: Meist autosomal-rezessiv. Es gibt auch Berichte über autosomal-dominante Vererbung.
Als Entstehungsmechanismen der angeborenen Lidkolobome sind unvollständiger Verschluss der Gesichtsspalten in der Embryonalzeit sowie Druck durch Amnionstränge bekannt.
QWie hängen verstecktes Auge und Fraser-Syndrom zusammen?
A
Bei 80–93 % der Patienten mit Fraser-Syndrom tritt ein verstecktes Auge auf, hauptsächlich verursacht durch Mutationen in den Genen FRAS1 und FREM2. Der FRAS/FREM-Komplex ist für die Aufrechterhaltung der Adhäsion zwischen Basalmembran und Epithel während der Embryonalentwicklung unerlässlich, und seine Mutation führt zu einer unvollständigen Trennung der Augenlider 1). Allerdings können FREM2-Mutationen auch die Ursache für ein isoliertes verstecktes Auge sein.
Pränataler Ultraschall kann etwa ab der 18. Schwangerschaftswoche nachweisen. Befunde sind das Fehlen der Lidspalte und eine durchgehende Haut vom Stirn bis zur Wange. Bei Vorliegen von Syndaktylie, erhöhter Lungenechogenität und Oligohydramnion ist ein Fraser-Syndrom wahrscheinlich.
Der FRAS/FREM-Panel-Test ist wichtig für die Diagnosebestätigung 1). Da die Phänotypen des Fraser-Syndroms und des MOTA-Syndroms überlappen, ist eine Unterscheidung allein anhand klinischer Befunde ohne Gentest oft schwierig.
Die Differenzialdiagnose umfasst angeborene Lidkolobome (bei teilweisem Defekt), Zyklopie und Anisokorie.
Die Behandlungsziele variieren je nach Typ. Bei der vollständigen und unvollständigen Form steht die ästhetische Rekonstruktion im Vordergrund, die Prognose für eine Sehverbesserung ist äußerst begrenzt. Bei der inkompletten Form ist die Behandlung des Risikos einer Hornhautexposition und Sehstörung dringend erforderlich.
Oberflächenmanagement des Auges: Verschreibung von ophthalmologischen Gleitmitteln und künstlichen Tränen (Maßnahmen gegen Exposition und Trockenheit). Bei großen Defekten wird eine Augensalbe zur Verhinderung von Hornhauttrockenheit verwendet.
Prothetische Augenabdeckung: Option, wenn eine Operation kontraindiziert, unmöglich oder erfolglos ist.
Schritt 1: Nach Hautinzision des Augenhöhlenrests wird ein mit Schleimhauttransplantat bedeckter Konformer eingesetzt, um den Bindehautsack zu bilden.
Schritt 2 (nach etwa einem Jahr): Lidrekonstruktion mit Verstärkung des hinteren Blatts und orbitaler Schleimhauttransplantation. Bei unzureichender Schleimhauttransplantation kann Vorhaut als Alternative verwendet werden.
Unvollständiger Typ / inkompletter Typ
Unvollständiger Typ: Nach Bildung eines Bindehautsacks (Schleimhauttransplantat + Conformer-Platzierung) erfolgt die Lidrekonstruktion mittels Lidteilung oder Switch-Methode. Es besteht ein Risiko für ein erneutes Auftreten von korneo-palpebralen Adhäsionen.
Inkompletter Typ: Hauptziel ist die Rekonstruktion des Oberlids und des oberen Fornix. Eine einzeitige Rekonstruktion mit Sklera- und Amniontransplantation wird durchgeführt.
Bei kleinen Defekten bei Kindern kann eine End-zu-End-Naht zur Reparatur ausreichen. Bei größeren Defekten wird eine plastisch-chirurgische Operation mit Hautlappen gewählt. Bei dem in der Fallstudie beschriebenen Kind zeigte die Augenexploration ein rudimentäres zystisches und adhärentes Auge; eine zusätzliche Operation wurde nicht durchgeführt1).
QWie wird die Operation bei vollständigem verstecktem Auge durchgeführt?
A
Sie wird schrittweise durchgeführt. Im ersten Schritt wird nach Hautinzision über dem Augenrest ein mit Schleimhauttransplantat bedeckter Konformer eingesetzt, um einen Bindehautsack zu schaffen. Etwa ein Jahr später erfolgt im zweiten Schritt die Lidrekonstruktion mit Verstärkung des hinteren Blatts und orbitaler Schleimhauttransplantation. Ziel ist hauptsächlich die Verbesserung des kosmetischen Erscheinungsbildes; die Prognose für eine Sehverbesserung ist äußerst selten.
6. Pathophysiologie und detaillierter Entstehungsmechanismus
Eine einheitliche Auffassung zur Pathophysiologie ist noch nicht etabliert.
Neuroektodermale Defekte: Die neuroektodermale Augenblase ist für die Entwicklung der fetalen Linse unerlässlich, und Defekte in dieser Schicht verhindern die ordnungsgemäße Entwicklung von Hornhaut, Linse und Vorderkammer.
Störungen der Lidbildung: Die Augenlider können sich ohne Differenzierung von Ektoderm und Mesoderm nicht bilden. Sie erscheinen in der 6. Embryonalwoche und sind bis zum 7. Monat verwachsen. Fehlbildungen in dieser Zeit führen zu angeborenen Lidfehlern.
Apoptose-Defekttheorie: Aufgrund des gleichzeitigen Auftretens von Syndaktylie, Kehlkopf- und Genitalanomalien wird angenommen, dass Defekte im programmierten Zelltod eine wichtige Rolle spielen.
Funktionsstörung des FRAS/FREM-Komplexes: Eine gestörte Adhäsion zwischen Basalmembran und Epithel durch FRAS1-, FREM1- und FREM2-Mutationen führt zu einer unvollständigen Lidspaltung1).
Zu den sekundären Komplikationen, die bei Mikrophthalmie häufig auftreten, gehören Katarakt, Linsenluxation, Glaukom und Netzhautablösung.
7. Aktuelle Forschung und zukünftige Perspektiven (Berichte aus der Forschung)
Mwipopo et al. (2023) berichteten über einen Fall von bilateralem Kryptophthalmus aus Tansania1). Gentests identifizierten eine FREM2-Mutation (heterozygot, likely pathogenic) sowie eine zusätzliche CEP85L-Mutation (Lissenzephalie 10: LIS10). LIS10 zeigt eine Heterogenität von leichter geistiger Behinderung bis hin zu schweren Phänotypen. Dieser Fall ist der erste Bericht aus Afrika, und es wurde darauf hingewiesen, dass die Nichtmeldung in Ländern mit niedrigem und mittlerem Einkommen zur Unterschätzung der Fallzahlen beiträgt.
Da Mutationen in Genen des FRAS/FREM-Komplexes bei mehreren Syndromen überlappende Phänotypen zeigen, ist eine rein klinische Diagnose unzureichend, und die Rolle von Gentests nimmt zu1). Auch in Ländern mit niedrigem und mittlerem Einkommen steigt der Bedarf an Gentests1).
Mwipopo E, Massomo MM, Moshiro R, Manji KP. Bilateral cryptophthalmos with overlapping features of Manitoba oculo-tricho-anal (MOTA) syndrome and Fraser syndrome 2. BMJ Case Rep. 2023;16(7):e252618.
Thomas IT, Frias JL, Felix V, Sanchez de Leon L, Hernandez RA, Jones MC. Isolated and syndromic cryptophthalmos. Am J Med Genet. 1986;25(1):85-98. PMID: 3099574.
Kabra M, Gulati S, Ghosh M, Menon PS. Fraser-cryptophthalmos syndrome. Indian J Pediatr. 2000;67(10):775-8. PMID: 11105430.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.