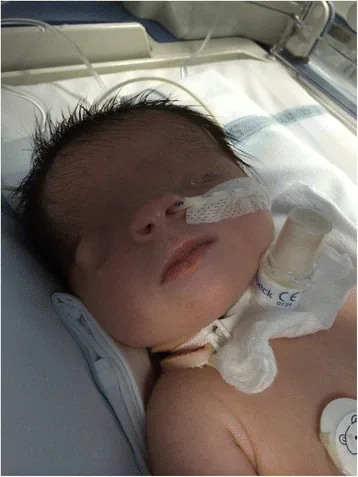
Cryptophthalmos berarti “mata tersembunyi” dalam bahasa Latin. Ini merujuk pada kondisi di mana kulit kelopak mata yang menyatu menutupi bola mata dan rongga mata, dengan tidak adanya celah kelopak mata. Kulit dahi menyatu dengan kulit pipi.
Angka kejadian dilaporkan 0,043 per 10.000 kelahiran hidup dan 1,1 per 10.000 lahir mati1). Hingga tahun 2018, hanya 55 kasus yang dilaporkan dalam literatur. Prevalensi keseluruhan kelainan kelopak mata adalah 0,06%, dengan 2/3 kasus sporadis dan 1/3 memiliki faktor genetik1).
Cryptophthalmos diklasifikasikan menjadi tiga tipe berdasarkan morfologi.
Tipe lengkap (tipikal): Oklusi total orbita. Tipe paling parah dengan tidak adanya alis, bulu mata, dan struktur kelenjar.
Tipe tidak lengkap (atipikal): Kelopak mata vestigial tersisa. Terdapat kantung konjungtiva kecil di sisi lateral.
Tipe abortif (bentuk abortif / sinekia palpebro-okular kongenital): Tidak adanya kelopak mata atas. Kulit dahi melekat pada bagian atas kornea.
Semua tipe dapat unilateral atau bilateral, terisolasi atau sindromik. Mata tersembunyi adalah mata yang tertutup oleh kulit kelopak mata yang menyatu, dapat muncul sendiri atau sebagai salah satu gejala sindrom Fraser. Jika tepi kelopak mata tidak terbentuk sama sekali, didiagnosis sebagai anoftalmia atau mata tersembunyi.
Sangat erat kaitannya dengan sindrom Fraser (Fraser syndrome), dan sebagian besar merupakan pewarisan autosomal resesif akibat mutasi gen FRAS1, GRIP1, dan FREM2. Cryptophthalmos ditemukan pada 80-93% kasus sindrom Fraser.
QDapatkah penglihatan anak dengan cryptophthalmos dipulihkan?
A
Hampir pada semua kasus, tidak ada atau sangat sedikit penglihatan. Tujuan operasi terutama untuk perbaikan penampilan kosmetik dan rekonstruksi area mata, sangat jarang perbaikan penglihatan dapat dicapai. Lihat bagian “Metode Pengobatan Standar” untuk detailnya.
De Bernardo G, Giordano M, Di Toro A, et al. Prenatal diagnosis of Fraser syndrome: a matter of life or death? Ital J Pediatr. 2015 Nov 9;41:86. Figure 1. PMCID: PMC4640198. License: CC BY.
Foto wajah segera setelah lahir, celah kelopak mata tidak terbentuk dan kulit menutupi permukaan mata. Mata kiri mikrofthalmos, dan mata kanan juga memiliki kelainan bentuk, membantu memahami penampilan cryptophthalmos.
Berikut adalah temuan klinis utama berdasarkan tipe.
Tipe lengkap
Oklusi orbita: Kulit dahi dan pipi bersambung, fisura palpebra sama sekali tidak ada.
Tidak ada adneksa: Alis, bulu mata, dan struktur kelenjar tidak ada.
Komplikasi mikroftalmia: Kulit permukaan menyatu dengan kornea. Tidak ada kantung konjungtiva, mikroftalmia sangat umum. Kista orbita dapat ditemukan.
Tipe tidak lengkap dan tipe parsial
Tipe tidak lengkap: Kelopak mata vestigial tersisa. Panjang fisura palpebra sekitar 1/3 normal. Terdapat kantung konjungtiva kecil di sisi lateral. Bola mata kecil dan hampir tertutup kulit.
Tipe tidak lengkap: Tidak ada kelopak mata atas. Kulit dahi menempel pada 75% bagian atas kornea. Kornea yang tertutup menjadi keratin dan keruh, tetapi kornea yang terbuka mungkin tetap jernih.
Dalam laporan kasus, seorang bayi laki-laki usia kehamilan 39 minggu ditemukan memiliki cryptophthalmos bilateral, tidak ada alis, garis rambut depan tidak teratur, celah ujung hidung, daun telinga rendah, hipertelorisme, dan malformasi anorektal rendah 1). Teraba kista orbita bergerak berukuran 1×1 cm pada mata kanan, dan mata kiri tertanam dalam 1). CT menunjukkan pola gyri abnormal di oksipital dan mata kiri yang mengecil 1).
Penyebab cryptophthalmos adalah mutasi gen pada protein yang membentuk kompleks FRAS/FREM.
Kompleks FRAS/FREM: Protein FRAS1, FREM1, dan FREM2 menjaga adhesi membran basal dengan epitel selama perkembangan embrio. Mutasi menyebabkan gangguan adhesi, mengakibatkan kegagalan pemisahan kelopak mata 1).
Sindrom Fraser (FRASRS1/2): Warisan autosomal resesif akibat mutasi gen FRAS1 atau FREM2 1).
Sindrom MOTA: Disebabkan oleh mutasi FREM1, ditandai dengan celah hidung, malformasi anorektal, agenesis ginjal, dan lainnya 1).
Pola pewarisan: Seringkali autosomal resesif. Ada juga laporan tentang autosomal dominan.
Mekanisme terjadinya defek kelopak mata bawaan meliputi kegagalan penutupan celah wajah pada masa embrio dan tekanan dari pita amnion.
QBagaimana hubungan antara cryptophthalmos dan sindrom Fraser?
A
Cryptophthalmos ditemukan pada 80-93% kasus sindrom Fraser, dan mutasi gen FRAS1/FREM2 merupakan penyebab utama. Kompleks FRAS/FREM sangat penting untuk mempertahankan adhesi membran basal dan epitel selama perkembangan embrio, dan mutasinya menyebabkan kegagalan pemisahan kelopak mata 1). Namun, mutasi FREM2 juga dapat menyebabkan cryptophthalmos terisolasi.
Dapat dideteksi dengan USG prenatal sekitar usia kehamilan 18 minggu. Temuannya berupa tidak adanya celah kelopak mata dan kulit yang kontinu dari dahi hingga pipi. Jika ditemukan sindaktili, peningkatan ekogenisitas paru, dan oligohidramnion, kemungkinan besar adalah sindrom Fraser.
Panel FRAS/FREM penting untuk memastikan diagnosis1). Karena fenotipe sindrom Fraser dan sindrom MOTA tumpang tindih, sulit membedakan hanya berdasarkan temuan klinis tanpa pemeriksaan genetik.
Diagnosis banding meliputi anoftalmia kongenital (jika defek parsial), siklopia, dan anisophthalmia.
Tujuan pengobatan berbeda tergantung tipe. Pada tipe komplit dan inkomplit, tujuan utama adalah rekonstruksi kosmetik, dan prognosis perbaikan penglihatan sangat terbatas. Pada tipe abortif, penanganan risiko keratopati eksposur dan gangguan penglihatan menjadi prioritas.
Manajemen permukaan mata: Resep pelumas mata dan air mata buatan (untuk mengatasi eksposur dan kekeringan). Jika defek besar, gunakan salep mata untuk mencegah kekeringan kornea.
Prostesis palpebra: Pilihan jika operasi kontraindikasi, tidak mungkin, atau gagal.
Tahap 1: Setelah sayatan kulit pada sisa bola mata, dimasukkan konformer yang dilapisi cangkok mukosa untuk membuat kantung konjungtiva.
Tahap 2 (sekitar 1 tahun kemudian): Dilakukan rekonstruksi kelopak mata dengan penguatan lamela posterior dan transplantasi mukosa orbita. Jika cangkok mukosa gagal, kulit kulup dapat digunakan sebagai alternatif.
Tipe Tidak Lengkap dan Tidak Sempurna
Tipe Tidak Lengkap: Setelah pembuatan kantung konjungtiva (cangkok mukosa + penempatan konformer), rekonstruksi kelopak mata dilakukan dengan bedah berbagi kelopak atau metode switch. Ada risiko rekurensi perlengketan kelopak mata-kornea.
Tipe tidak lengkap: Tujuan utama adalah rekonstruksi kelopak mata atas dan forniks superior. Rekonstruksi satu tahap dilakukan dengan transplantasi sklera atau amnion.
Jika defek pada anak kecil, dapat diperbaiki dengan jahitan ujung ke ujung. Jika besar, operasi plastik dengan flap kulit dipilih. Pada kasus yang dilaporkan, ditemukan bola mata kistik rudimenter yang melekat saat eksplorasi, dan tidak dilakukan operasi tambahan1).
QBagaimana operasi untuk mata tersembunyi total dilakukan?
A
Dilakukan secara bertahap. Tahap pertama, setelah sayatan kulit pada sisa bola mata, dimasukkan konformer yang dilapisi cangkok mukosa untuk membuat kantung konjungtiva. Sekitar satu tahun kemudian, pada tahap kedua, dilakukan rekonstruksi kelopak mata dengan penguatan lamela posterior dan cangkok mukosa orbita. Tujuan utamanya adalah perbaikan penampilan kosmetik, dan prognosis perbaikan penglihatan sangat jarang.
Belum ada konsensus yang mapan mengenai patofisiologi.
Defek neuroektodermal: Vesikel optik neuroektodermal penting untuk perkembangan lensa janin, dan defek pada lapisan ini menghambat perkembangan kornea, lensa, dan bilik mata depan yang tepat.
Gangguan pembentukan kelopak mata: Kelopak mata tidak terbentuk tanpa diferensiasi ektoderm dan mesoderm. Kelopak mata muncul pada minggu ke-6 kehamilan, dan kelopak atas dan bawah menyatu hingga bulan ke-7. Kegagalan pembentukan selama periode ini menyebabkan kelainan kelopak mata bawaan.
Teori defek apoptosis: Adanya sindaktili, kelainan laring, dan genital menunjukkan bahwa defek apoptosis memainkan peran penting.
Disfungsi kompleks FRAS/FREM: Gangguan adhesi membran basal dan epitel akibat mutasi FRAS1, FREM1, dan FREM2 menyebabkan kegagalan pemisahan kelopak mata1).
Komplikasi sekunder yang sering menyertai mikroftalmia meliputi katarak, dislokasi lensa, glaukoma, dan ablasi retina.
7. Penelitian terbaru dan prospek ke depan (laporan tahap penelitian)
Mwipopo dkk. (2023) melaporkan kasus mata tersembunyi bilateral dari Tanzania 1). Pemeriksaan genetik mengidentifikasi mutasi FREM2 (kemungkinan patogenik heterozigot) bersama dengan mutasi CEP85L (lissencephaly 10: LIS10). LIS10 menunjukkan heterogenitas dari keterbelakangan mental ringan hingga fenotip berat. Kasus ini merupakan laporan pertama dari Afrika, dan disebutkan bahwa kurangnya pelaporan di negara berpenghasilan rendah dan menengah berkontribusi pada underestimasi jumlah kasus.
Karena mutasi gen terkait kompleks FRAS/FREM menunjukkan fenotip yang tumpang tindih pada beberapa sindrom, diagnosis klinis saja tidak cukup, dan peran pemeriksaan genetik semakin meningkat 1). Kebutuhan akan pemeriksaan genetik juga meningkat di negara berpenghasilan rendah dan menengah 1).
Mwipopo E, Massomo MM, Moshiro R, Manji KP. Bilateral cryptophthalmos with overlapping features of Manitoba oculo-tricho-anal (MOTA) syndrome and Fraser syndrome 2. BMJ Case Rep. 2023;16(7):e252618.
Thomas IT, Frias JL, Felix V, Sanchez de Leon L, Hernandez RA, Jones MC. Isolated and syndromic cryptophthalmos. Am J Med Genet. 1986;25(1):85-98. PMID: 3099574.
Kabra M, Gulati S, Ghosh M, Menon PS. Fraser-cryptophthalmos syndrome. Indian J Pediatr. 2000;67(10):775-8. PMID: 11105430.
Salin teks artikel dan tempelkan ke asisten AI pilihan Anda.
Artikel disalin ke papan klip
Buka asisten AI di bawah, lalu tempelkan teks yang disalin ke kotak chat.