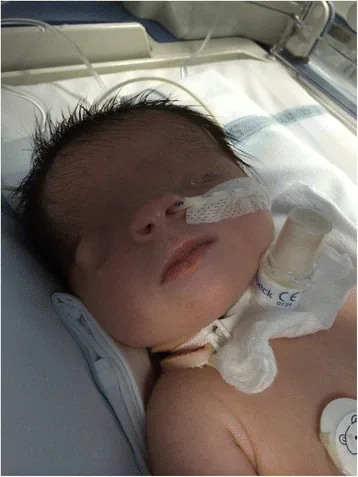
Criptoftalmos significa “ojo oculto” en latín. Se refiere a una condición en la que la piel del párpado fusionada cubre el globo ocular y la órbita, con ausencia de la hendidura palpebral. La piel de la frente es continua con la piel de la mejilla.
La incidencia se reporta como 0.043 por cada 10,000 nacidos vivos y 1.1 por cada 10,000 mortinatos1). Hasta 2018, solo se han reportado 55 casos en la literatura. La prevalencia general de anomalías palpebrales es del 0.06%, con dos tercios esporádicos y un tercio con predisposición genética1).
El criptoftalmos se clasifica en tres tipos según la morfología.
Completo (típico): Oclusión completa de la órbita. El tipo más grave, con ausencia de cejas, pestañas y estructuras glandulares.
Incompleto (atípico): Persisten párpados rudimentarios. Hay un pequeño saco conjuntival en el lado lateral.
Forma abortiva (simbelfarón congénito): Ausencia del párpado superior. La piel de la frente se adhiere a la parte superior de la córnea.
Cualquier tipo puede ser unilateral o bilateral, aislado o sindrómico. El criptoftalmos es una condición en la que el globo ocular está cubierto por piel palpebral fusionada, y puede aparecer solo o como parte del síndrome de Fraser. Si el borde del párpado no está formado en absoluto, se diagnostica como ablefaria o criptoftalmos.
Está muy estrechamente asociado con el síndrome de Fraser, y es común la herencia autosómica recesiva debida a mutaciones en los genes FRAS1, GRIP1 y FREM2. Se observa criptoftalmos en el 80–93% de los casos de síndrome de Fraser.
Q¿Puede un niño con criptoftalmos recuperar la visión?
A
En casi todos los casos, hay poca o ninguna función visual. El propósito de la cirugía es principalmente la mejora estética y la reconstrucción de la región ocular; la mejora de la visión es extremadamente rara. Para más detalles, consulte la sección sobre “Tratamientos estándar”.
De Bernardo G, Giordano M, Di Toro A, et al. Prenatal diagnosis of Fraser syndrome: a matter of life or death? Ital J Pediatr. 2015 Nov 9;41:86. Figure 1. PMCID: PMC4640198. License: CC BY.
Una fotografía facial tomada poco después del nacimiento muestra que la hendidura palpebral no se ha formado y la piel cubre la superficie ocular. El ojo izquierdo es microftálmico, y el ojo derecho también presenta anomalías morfológicas, lo que ayuda a comprender la apariencia del criptoftalmos.
Los principales hallazgos clínicos por tipo se muestran a continuación.
Tipo Completo
Oclusión orbitaria: La piel de la frente y la mejilla es continua, con ausencia completa de la hendidura palpebral.
Ausencia de anexos: Las cejas, pestañas y estructuras glandulares están ausentes.
Asociación con microftalmía: La piel superficial se fusiona con la córnea. No hay saco conjuntival, y la microftalmía es muy común. Pueden existir quistes orbitarios.
Tipo Incompleto / Parcial
Tipo incompleto: Quedan párpados rudimentarios. La longitud de la hendidura palpebral es aproximadamente un tercio de lo normal. Hay un pequeño saco conjuntival lateralmente. El globo ocular es pequeño y está casi cubierto por la piel.
Tipo incompleto: Ausencia del párpado superior. La piel de la frente se adhiere al 75% superior de la córnea. La córnea cubierta se queratiniza y opacifica, pero la córnea expuesta puede ser transparente.
En un reporte de caso, un varón de 39 semanas de gestación presentó criptoftalmos bilateral, ausencia de cejas, desorden de la línea del cabello frontal, hendidura de la punta nasal, orejas bajas, hipertelorismo y malformación anorrectal baja1). Se palpó un quiste orbitario móvil de 1×1 cm en el ojo derecho, y el globo ocular izquierdo estaba profundamente enterrado1). La TC reveló un patrón anormal de giros en la región occipital y un globo ocular izquierdo pequeño1).
El criptoftalmos es causado por mutaciones genéticas en proteínas que forman el complejo FRAS/FREM.
Complejo FRAS/FREM: Las proteínas FRAS1, FREM1 y FREM2 mantienen la adhesión entre la membrana basal y el epitelio durante el desarrollo embrionario. Las mutaciones causan defectos de adhesión, lo que lleva a una falla en la separación palpebral1).
Síndrome de Fraser (FRASRS1/2): Herencia autosómica recesiva debida a mutaciones en los genes FRAS1 o FREM21).
Síndrome MOTA: Mutación de FREM1 asociada con hendidura nasal, malformaciones anorrectales, agenesia renal, etc. 1).
Criptoftalmos aislado: La mutación de FREM2 también puede causar criptoftalmos aislado unilateral o bilateral 1).
Patrón de herencia: Mayormente autosómico recesivo. También se ha reportado herencia autosómica dominante.
Los mecanismos del coloboma palpebral congénito incluyen el cierre incompleto de las hendiduras faciales durante el desarrollo embrionario y la compresión por bandas amnióticas.
Q¿Cómo se relaciona el criptoftalmos con el síndrome de Fraser?
A
El criptoftalmos está presente en el 80–93% de los casos de síndrome de Fraser, y las mutaciones en FRAS1 y FREM2 son la causa principal. El complejo FRAS/FREM es esencial para mantener la adhesión entre la membrana basal y el epitelio durante el desarrollo embrionario, y su mutación provoca la falta de separación de los párpados 1). Sin embargo, las mutaciones en FREM2 también pueden causar criptoftalmos aislado.
La ecografía prenatal puede detectarlo alrededor de las 18 semanas de gestación. Los hallazgos incluyen ausencia de la hendidura palpebral y piel continua desde la frente hasta la mejilla. Si se presentan sindactilia, aumento de la ecogenicidad pulmonar y oligohidramnios, es muy probable que sea síndrome de Fraser.
Los criterios diagnósticos para el síndrome de Fraser se muestran a continuación.
Clasificación
Ítem
Criterios mayores
Criptoftalmos, sindactilia, anomalías genitales, anomalías de extremidades
Criterios menores
Deformidades de oreja/nariz, labio/paladar hendido, línea del cabello anormal, malformaciones renales
Pruebas necesarias:
Examen de agudeza visual, estrabismo y fondo de ojo: Evaluación de la función visual
Prueba de tracción bajo anestesia general: Confirmación de posibles bandas fibrosas
TC/RM: Evaluación de la morfología ocular y el cerebro. Útil para confirmar complicaciones del sistema nervioso central, como patrones anormales de giros occipitales 1)
La prueba del panel FRAS/FREM es importante para el diagnóstico definitivo 1). Debido a que los fenotipos del síndrome de Fraser y del síndrome MOTA se superponen, la diferenciación basada únicamente en hallazgos clínicos sin pruebas genéticas puede ser difícil.
El diagnóstico diferencial incluye coloboma palpebral congénito (en casos de defecto parcial), ciclopía y anisoftalmia.
Los objetivos del tratamiento varían según el tipo. En los tipos completo e incompleto, la reconstrucción cosmética es el objetivo principal y el pronóstico de mejora visual es extremadamente limitado. En el tipo abortivo, es urgente abordar la queratopatía por exposición y el riesgo de deterioro visual.
Manejo de la superficie ocular: Prescripción de lubricantes oftálmicos y lágrimas artificiales (para exposición y sequedad). Para defectos grandes, use ungüento oftálmico para prevenir la sequedad corneal.
Cáscara protésica: Opción cuando la cirugía está contraindicada, es imposible o no tiene éxito.
Etapa 1: Después de la incisión cutánea del remanente ocular, se inserta un conformador cubierto con un injerto de mucosa para crear un saco conjuntival.
Etapa 2 (aproximadamente 1 año después): Se realiza la reconstrucción palpebral con refuerzo de la lámina posterior e injerto de mucosa orbitaria. Si el injerto de mucosa no es efectivo, se puede usar el prepucio como alternativa.
Tipos incompleto y parcial
Tipo incompleto: Después de la creación del saco conjuntival (injerto de mucosa + colocación del conformador), se realiza la reconstrucción palpebral mediante la técnica de compartir párpado o colgajo de intercambio. Existe riesgo de recurrencia de la adherencia córneo-palpebral.
Tipo incompleto: El objetivo principal es la reconstrucción del párpado superior y del fondo de saco superior. Se realiza una reconstrucción en un solo tiempo mediante injerto de esclerótica y membrana amniótica.
Si el defecto en un niño es pequeño, se puede reparar mediante sutura término-terminal. Si es grande, se opta por cirugía plástica con colgajos cutáneos. En el caso reportado, la exploración ocular reveló un globo ocular rudimentario quístico y adherido, y no se realizó cirugía adicional1).
Q¿Cómo se realiza la cirugía para el criptoftalmos completo?
A
Se realiza por etapas. En la primera etapa, después de una incisión cutánea sobre el remanente ocular, se inserta un conformador cubierto con un injerto de mucosa para crear un saco conjuntival. Aproximadamente un año después, en la segunda etapa, se realiza la reconstrucción palpebral con refuerzo de la lámina posterior e injerto de mucosa orbitaria. El objetivo principal es la mejora cosmética; la mejora visual es extremadamente rara.
Aún no se ha establecido un consenso sobre la fisiopatología.
Defecto neuroectodérmico: La vesícula óptica neuroectodérmica es esencial para el desarrollo del cristalino fetal, y los defectos en esta capa impiden el desarrollo adecuado de la córnea, el cristalino y la cámara anterior.
Alteración de la formación palpebral: Los párpados no pueden formarse sin la diferenciación del ectodermo y el mesodermo. Los párpados aparecen a las 6 semanas de gestación, y los párpados superior e inferior están fusionados hasta el séptimo mes de gestación. La malformación durante este período conduce a anomalías palpebrales congénitas.
Teoría del defecto de apoptosis: La combinación de sindactilia, anomalías laríngeas y genitales sugiere que un defecto en la muerte celular programada juega un papel importante.
Disfunción del complejo FRAS/FREM: La alteración de la adhesión entre la membrana basal y el epitelio debido a mutaciones en FRAS1, FREM1 y FREM2 provoca la falta de separación palpebral 1).
Las complicaciones secundarias que a menudo acompañan a la microftalmía incluyen cataratas, luxación del cristalino, glaucoma y desprendimiento de retina.
7. Investigación más reciente y perspectivas futuras (Informes en fase de investigación)
Mwipopo et al. (2023) reportaron un caso de criptoftalmos bilateral de Tanzania 1). Las pruebas genéticas identificaron una mutación en FREM2 (heterocigota probablemente patogénica) junto con una mutación en CEP85L (lisencefalia 10: LIS10). LIS10 muestra heterogeneidad que va desde discapacidad intelectual leve hasta fenotipos graves. Este caso es el primer informe de África, y se señaló que la falta de notificación en países de ingresos bajos y medios contribuye a la subestimación del número de casos.
Dado que las mutaciones en genes relacionados con el complejo FRAS/FREM muestran fenotipos superpuestos en múltiples síndromes, el diagnóstico clínico por sí solo es insuficiente y el papel de las pruebas genéticas está aumentando 1). La necesidad de pruebas genéticas también está creciendo en países de ingresos bajos y medios 1).
Mwipopo E, Massomo MM, Moshiro R, Manji KP. Bilateral cryptophthalmos with overlapping features of Manitoba oculo-tricho-anal (MOTA) syndrome and Fraser syndrome 2. BMJ Case Rep. 2023;16(7):e252618.
Thomas IT, Frias JL, Felix V, Sanchez de Leon L, Hernandez RA, Jones MC. Isolated and syndromic cryptophthalmos. Am J Med Genet. 1986;25(1):85-98. PMID: 3099574.
Kabra M, Gulati S, Ghosh M, Menon PS. Fraser-cryptophthalmos syndrome. Indian J Pediatr. 2000;67(10):775-8. PMID: 11105430.
Copia el texto del artículo y pégalo en el asistente de IA que prefieras.
Artículo copiado al portapapeles
Abre un asistente de IA abajo y pega el texto copiado en el chat.