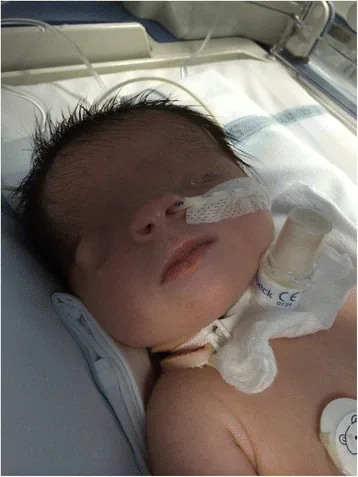
Kriptoftalmi, Latince’de “gizli göz” anlamına gelir. Kaynaşmış göz kapağı derisinin göz küresini ve yuvasını kaplaması, palpebral fissürün olmaması durumunu ifade eder. Alın derisi yanak derisi ile sürekli olarak oluşur.
Görülme sıklığı canlı doğumlarda 10.000’de 0.043, ölü doğumlarda 10.000’de 1.1 olarak bildirilmiştir1). 2018 itibarıyla literatürde yalnızca 55 vaka rapor edilmiştir. Tüm göz kapağı anomalilerinin prevalansı %0.06 olup, 2/3’ü sporadik, 1/3’ü genetik yatkınlık gösterir1).
Komplet (tipik) tip: Orbital bölgenin tamamen kapanması. Kaş, kirpik ve bez yapılarının yokluğu ile en şiddetli form.
İnkomplet (atipik) tip: Kalıntı göz kapakları mevcuttur. Lateralde küçük bir konjonktival kese bulunur.
Eksik form (abortive form / konjenital blefarosinekşi): Üst göz kapağı yokluğu. Alın derisi korneanın üst kısmına yapışır.
Her iki tip de tek taraflı veya iki taraflı, izole veya sendromik olabilir. Gizli göz, kaynaşmış göz kapağı derisiyle kaplı göz küresidir; tek başına veya Fraser sendromunun bir belirtisi olarak ortaya çıkabilir. Göz kapağı kenarı tamamen oluşmamışsa, anoftalmi veya gizli göz tanısı konur.
Fraser sendromu ile çok yakından ilişkilidir; FRAS1, GRIP1 ve FREM2 gen mutasyonlarına bağlı otozomal resesif kalıtım sıktır. Fraser sendromlu hastaların %80-93’ünde kriptoftalmi görülür.
QKriptoftalmili bir çocuğun görme yetisi geri kazanılabilir mi?
A
Hemen hemen tüm vakalarda görme yetisi yok denecek kadar azdır veya hiç yoktur. Cerrahinin amacı esas olarak kozmetik görünümü iyileştirmek ve göz bölgesini rekonstrükte etmektir; görme keskinliğinde iyileşme son derece nadirdir. Ayrıntılar için “Standart tedavi yöntemleri” bölümüne bakın.
De Bernardo G, Giordano M, Di Toro A, et al. Prenatal diagnosis of Fraser syndrome: a matter of life or death? Ital J Pediatr. 2015 Nov 9;41:86. Figure 1. PMCID: PMC4640198. License: CC BY.
Doğumdan hemen sonra çekilen yüz fotoğrafında, göz aralığının oluşmadığı ve derinin göz yüzeyini kapladığı görülüyor. Solda mikrof talmus, sağ gözde de şekil bozukluğu mevcut; bu, kriptoftalmus görünümünü anlamaya yardımcı olur.
Tipe göre başlıca klinik bulgular aşağıda gösterilmiştir.
Komplet Tip
Orbital tıkanıklık: Alın ve yanak derisi süreklidir, palpebral fissür tamamen yoktur.
Eklere ait yokluk: Kaş, kirpik ve bez yapıları mevcut değildir.
Mikroftalmi birlikteliği: Yüzey derisi kornea ile kaynaşmıştır. Konjonktival kese yoktur, mikroftalmi çok yaygındır. Orbital kistler bulunabilir.
İnkomplet tip
İnkomplet tip: Kalıntı göz kapağı mevcuttur. Palpebral fissür uzunluğu normalin yaklaşık 1/3’ü kadardır. Lateralde küçük bir konjonktival kese vardır. Göz küresi küçüktür ve neredeyse tamamen deri ile kaplıdır.
İnkomplet tip: Üst göz kapağı yokluğu. Alın derisi korneanın üst %75’ine yapışıktır. Örtülü kornea keratinize ve bulanık olabilir, ancak açıkta kalan kornea saydam olabilir.
Vaka raporunda, 39. gebelik haftasında doğan bir erkek bebekte bilateral kriptoftalmi, kaş yokluğu, frontal saç çizgisinde düzensizlik, burun ucu yarığı, düşük kulak kepçeleri, hipertelorizm, düşük anorektal malformasyon bildirilmiştir1). Sağ gözde 1×1 cm boyutunda hareketli orbital kist palpe edilmiş, sol göz küresi derin bir şekilde gömülüydü1). BT’de oksipital bölgede anormal girus paterni ve sol göz küresinde küçülme saptanmıştır1).
Kriptoftalminin nedeni, FRAS/FREM kompleksini oluşturan proteinlerdeki gen mutasyonlarıdır.
FRAS/FREM kompleksi: FRAS1, FREM1 ve FREM2 proteinleri embriyonik gelişim sırasında bazal membran ve epitel yapışmasını sürdürür. Mutasyonlar yapışma bozukluğuna yol açar ve göz kapağı ayrışmamasına neden olur1).
Fraser sendromu (FRASRS1/2): FRAS1 veya FREM2 gen mutasyonlarına bağlı otozomal resesif kalıtım1).
MOTA sendromu: FREM1 mutasyonu burun yarığı, anorektal malformasyon ve renal agenezi gibi bulgularla birlikte görülür1).
İzole kriptoftalmi: FREM2 mutasyonu tek taraflı veya iki taraflı izole kriptoftalminin de nedeni olabilir1).
Kalıtım şekli: Çoğunlukla otozomal resesif kalıtım. Otozomal dominant kalıtım da bildirilmiştir.
Konjenital göz kapağı kolobomunun oluşum mekanizması olarak, embriyonik dönemde yüz yarıklarının kapanma yetersizliği ve amniyotik bant basısı bilinmektedir.
QKriptoftalmi ve Fraser sendromu nasıl ilişkilidir?
A
Fraser sendromlu hastaların %80-93’ünde kriptoftalmi görülür ve başlıca neden FRAS1/FREM2 gen mutasyonlarıdır. FRAS/FREM kompleksi, embriyonik gelişim sırasında bazal membran ve epitel yapışmasının sürdürülmesi için gereklidir ve mutasyonu göz kapağı ayrışmasında bozukluğa yol açar1). Ancak FREM2 mutasyonu izole kriptoftalmiye de neden olabilir.
Prenatal ultrasonografi ile gebeliğin 18. haftası civarında tespit edilebilir. Palpebral fissür yokluğu ve alından yanağa kadar uzanan sürekli cilt bulgulardır. Sindaktili, akciğer ekojenitesinde artış ve oligohidramnios varlığında Fraser sendromu olasılığı yüksektir.
Görme keskinliği, şaşılık ve fundus muayenesi: Görme fonksiyonunun değerlendirilmesi
Genel anestezi altında traksiyon testi: Bant benzeri yapıların gizli varlığının kontrolü
BT/MRG: Göz küresi morfolojisi ve beyin değerlendirmesi. Oksipital bölgede anormal girus paterni gibi santral sinir sistemi komplikasyonlarının saptanmasında faydalıdır1)
FRAS/FREM panel testi tanıyı doğrulamak için önemlidir1). Fraser sendromu ve MOTA sendromunun fenotipleri örtüştüğü için, genetik test olmadan sadece klinik bulgularla ayırıcı tanı yapmak zor olabilir.
Ayırıcı tanıda konjenital göz kapağı kolobomu (kısmi defekt durumunda), siklopi ve anizoftalmi yer alır.
Tedavi hedefi tipe göre değişir. Komplet ve inkomplet tipte temel amaç kozmetik rekonstrüksiyondur ve görme iyileşmesi prognozu oldukça sınırlıdır. İnkomplet tipte ise maruziyet keratopatisi ve görme bozukluğu riskine acil müdahale gereklidir.
Oküler yüzey yönetimi: Oftalmik kayganlaştırıcılar ve suni gözyaşı reçetesi (maruziyet ve kuruluk önlemi). Defekt büyükse kornea kuruluğunu önlemek için göz merhemi kullanılır.
Protez göz kapağı: Cerrahinin kontrendike, mümkün olmadığı veya başarısız olduğu durumlarda seçenek.
1. Aşama: Göz küresi kalıntısının deri insizyonundan sonra, mukoza grefti ile kaplı bir konformer yerleştirilerek konjonktival kese oluşturulur.
2. Aşama (yaklaşık 1 yıl sonra): Arka lamella güçlendirme ve orbital mukoza grefti ile birlikte göz kapağı rekonstrüksiyonu yapılır. Mukoza grefti başarısız olursa, alternatif olarak sünnet derisi kullanılabilir.
İnkomplet Tip
İnkomplet tip: Konjonktival kese oluşturulması (mukozal greft + konformer yerleştirilmesi) ardından, tarsorafi veya switch yöntemi ile göz kapağı rekonstrüksiyonu yapılır. Kornea-göz kapağı yapışıklığı (sembelfaron) tekrarlama riski vardır.
Komplet tip: Üst göz kapağı ve üst forniks rekonstrüksiyonu ana hedeftir. Sklera ve amniyon membran grefti kullanılarak tek aşamalı rekonstrüksiyon yapılır.
Pediatrik defekt küçükse uç uca sütür ile onarılabilir. Büyükse deri flebi ile plastik cerrahi yöntemler tercih edilir. Olgu sunumundaki çocukta göz küresi araştırmasında kalıntı kistik ve yapışık göz küresi tespit edilmiş, ek cerrahi yapılmamıştır1).
QTam tip kriptoftalmi ameliyatı nasıl yapılır?
A
Aşamalı olarak gerçekleştirilir. İlk aşamada, göz küresi kalıntısının deri insizyonundan sonra mukoza grefti ile kaplı bir konformer yerleştirilerek konjonktival kese oluşturulur. Yaklaşık bir yıl sonraki ikinci aşamada, arka lamella güçlendirme ve orbital mukoza grefti ile birlikte göz kapağı rekonstrüksiyonu yapılır. Amaç esas olarak kozmetik görünümün iyileştirilmesidir; görme iyileşmesi prognozu son derece nadirdir.
Patofizyoloji ile ilgili ortak bir görüş henüz oluşturulmamıştır.
Nöroektodermal defekt: Nöroektodermal optik vezikül, fetal lens gelişimi için gereklidir ve bu tabakanın defekti, kornea, lens ve ön kamaranın uygun gelişimini engeller.
Göz kapağı oluşum bozukluğu: Göz kapakları, ektoderm ve mezoderm farklılaşması olmadan oluşamaz. Göz kapakları embriyonik 6. haftada belirir ve üst-alt göz kapakları embriyonik 7. aya kadar kaynaşık kalır. Bu dönemdeki gelişim kusurları konjenital göz kapağı anomalilerine yol açar.
Apoptoz defekti teorisi: Sindaktili, larenks ve genital anormalliklerin birlikteliği, programlı hücre ölümü defektinin önemli bir rol oynadığını düşündürmektedir.
FRAS/FREM kompleks disfonksiyonu: FRAS1, FREM1 ve FREM2 mutasyonlarına bağlı bazal membran ve epitel yapışma bozukluğu, göz kapağı ayrışma yetersizliğine yol açar1).
Mikroftalmiye sık eşlik eden ikincil komplikasyonlar arasında katarakt, lens dislokasyonu, glokom ve retina dekolmanı bulunur.
7. Güncel Araştırmalar ve Gelecek Perspektifleri (Araştırma Aşamasındaki Raporlar)
Mwipopo ve ark. (2023), Tanzanya’dan iki taraflı kriptoftalmi vakası bildirmiştir1). Genetik test, FREM2 mutasyonuna (heterozigot olası patojenik) ek olarak, CEP85L mutasyonunun (lizensefali 10: LIS10) birlikte bulunduğunu tanımlamıştır. LIS10, hafif zihinsel gerilikten şiddetli fenotipe kadar heterojenlik gösterir. Bu vaka, Afrika’dan ilk bildirilen vakadır ve düşük-orta gelirli ülkelerde bildirilmeyen vakaların, vaka sayısının eksik tahmin edilmesine katkıda bulunduğu belirtilmiştir.
FRAS/FREM kompleksinin ilişkili gen mutasyonları, birden fazla sendromda örtüşen fenotipler gösterdiğinden, yalnızca klinik tanı yeterli değildir ve genetik testin rolü artmaktadır1). Düşük-orta gelirli ülkelerde de genetik teste olan ihtiyaç artmaktadır1).
Mwipopo E, Massomo MM, Moshiro R, Manji KP. Bilateral cryptophthalmos with overlapping features of Manitoba oculo-tricho-anal (MOTA) syndrome and Fraser syndrome 2. BMJ Case Rep. 2023;16(7):e252618.
Thomas IT, Frias JL, Felix V, Sanchez de Leon L, Hernandez RA, Jones MC. Isolated and syndromic cryptophthalmos. Am J Med Genet. 1986;25(1):85-98. PMID: 3099574.
Kabra M, Gulati S, Ghosh M, Menon PS. Fraser-cryptophthalmos syndrome. Indian J Pediatr. 2000;67(10):775-8. PMID: 11105430.
Makale metnini kopyalayıp tercih ettiğiniz yapay zeka asistanına yapıştırabilirsiniz.
Makale panoya kopyalandı
Aşağıdaki yapay zeka asistanlarından birini açın ve kopyalanan metni sohbet kutusuna yapıştırın.