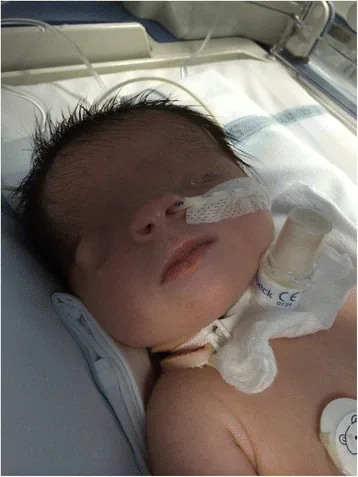
Le cryptophtalmos signifie « œil caché » en latin. Il désigne une condition où la peau palpébrale fusionnée recouvre le globe oculaire et l’orbite, avec absence de fente palpébrale. La peau du front est continue avec celle de la joue.
L’incidence est rapportée à 0,043 pour 10 000 naissances vivantes et 1,1 pour 10 000 mortinaissances1). En 2018, seulement 55 cas étaient documentés dans la littérature. La prévalence globale des anomalies palpébrales est de 0,06 %, dont 2/3 sont sporadiques et 1/3 présentent une prédisposition génétique1).
Le cryptophtalmos est classé en trois types selon la morphologie.
Type complet (typique) : occlusion complète de l’orbite. Forme la plus sévère avec absence de sourcils, de cils et de structures glandulaires.
Type incomplet (atypique) : présence de paupières rudimentaires. Petit cul-de-sac conjonctival latéral.
Forme abortive (abortive form / adhérence congénitale palpébro-oculaire) : Absence de paupière supérieure. La peau du front adhère à la partie supérieure de la cornée.
Tous les types peuvent être unilatéraux ou bilatéraux, isolés ou syndromiques. Le cryptophtalmos est un œil recouvert par une peau palpébrale fusionnée, pouvant apparaître isolément ou comme l’un des symptômes du syndrome de Fraser. Si le bord palpébral n’est pas du tout formé, on diagnostique une absence de paupière ou un cryptophtalmos.
Le lien avec le syndrome de Fraser est très étroit, avec une transmission autosomique récessive fréquente due à des mutations des gènes FRAS1, GRIP1 et FREM2. Le cryptophtalmos est observé dans 80 à 93 % des cas de syndrome de Fraser.
QUn enfant atteint de cryptophtalmos peut-il retrouver la vue ?
A
Dans presque tous les cas, la vision est absente ou très limitée. Le but de la chirurgie est principalement d’améliorer l’apparence esthétique et de reconstruire la région orbitaire ; une amélioration de la vision est extrêmement rare. Voir la section « Traitement standard » pour plus de détails.
De Bernardo G, Giordano M, Di Toro A, et al. Prenatal diagnosis of Fraser syndrome: a matter of life or death? Ital J Pediatr. 2015 Nov 9;41:86. Figure 1. PMCID: PMC4640198. License: CC BY.
Photographie du visage juste après la naissance montrant que la fente palpébrale n’est pas formée et que la peau recouvre la surface oculaire. L’œil gauche est microphtalme et l’œil droit présente également des anomalies morphologiques, ce qui aide à comprendre l’apparence de la cryptophtalmie.
Les principaux signes cliniques selon le type sont présentés ci-dessous.
Type complet
Obstruction orbitaire : la peau du front et de la joue est continue, et la fente palpébrale est totalement absente.
Absence des annexes : Les sourcils, les cils et les structures glandulaires sont absents.
Association avec une microphtalmie : La peau superficielle fusionne avec la cornée. Il n’y a pas de cul-de-sac conjonctival, et la microphtalmie est très fréquente. Des kystes orbitaires peuvent être présents.
Forme incomplète
Forme incomplète : Des vestiges de paupières persistent. La fente palpébrale mesure environ un tiers de la longueur normale. Il existe un petit cul-de-sac conjonctival latéral. Le globe oculaire est petit et presque entièrement recouvert par la peau.
Forme incomplète : absence de la paupière supérieure. La peau du front adhère à 75 % de la partie supérieure de la cornée. La cornée recouverte devient kératinisée et trouble, mais la cornée exposée peut rester transparente.
Dans un rapport de cas, un garçon né à 39 semaines de gestation présentait une cryptophtalmie bilatérale, une absence de sourcils, une ligne capillaire frontale irrégulière, une fente nasale, des oreilles basses, un hypertélorisme et une malformation anorectale basse1). Un kyste orbitaire mobile de 1×1 cm était palpable à l’œil droit, et le globe oculaire gauche était profondément enfoui1). Le scanner a révélé un motif anormal de gyri occipitaux et une réduction du globe oculaire gauche1).
La cryptophtalmie est causée par des mutations génétiques des protéines constituant le complexe FRAS/FREM.
Complexe FRAS/FREM : les protéines FRAS1, FREM1 et FREM2 maintiennent l’adhésion entre la membrane basale et l’épithélium au cours du développement embryonnaire. Les mutations entraînent un défaut d’adhésion, provoquant une cryptophtalmie1).
Syndrome de Fraser (FRASRS1/2) : maladie autosomique récessive due à des mutations des gènes FRAS1 ou FREM21).
Syndrome MOTA : les mutations de FREM1 s’accompagnent de fente nasale, de malformations anorectales et d’agénésie rénale1).
Cryptophtalmie isolée : les mutations de FREM2 peuvent également être à l’origine d’une cryptophtalmie isolée unilatérale ou bilatérale1).
Mode de transmission : le plus souvent autosomique récessif. Des cas autosomiques dominants ont également été rapportés.
Les mécanismes à l’origine de la colobome palpébral congénital incluent une fermeture incomplète des fentes faciales au cours de la période embryonnaire et une compression par des brides amniotiques.
QQuel est le lien entre le cryptophtalmos et le syndrome de Fraser ?
A
Le cryptophtalmos est présent dans 80 à 93 % des cas de syndrome de Fraser, principalement dû à des mutations des gènes FRAS1 et FREM2. Le complexe FRAS/FREM est essentiel au maintien de l’adhésion entre la membrane basale et l’épithélium au cours du développement embryonnaire, et sa mutation entraîne une non-séparation des paupières 1). Cependant, les mutations de FREM2 peuvent également être à l’origine d’un cryptophtalmos isolé.
L’échographie prénatale permet de détecter la maladie vers la 18e semaine de grossesse. Les signes sont l’absence de fente palpébrale et une peau continue du front à la joue. La présence de syndactylie, d’hyperéchogénicité pulmonaire et d’oligohydramnios suggère fortement le syndrome de Fraser.
Les critères diagnostiques du syndrome de Fraser sont présentés ci-dessous.
Catégorie
Élément
Critère principal
Cryptophtalmie, syndactylie, anomalies génitales, anomalies des membres
Critère secondaire
Déformations des oreilles et du nez, fente labiopalatine, anomalie de la ligne des cheveux, malformation rénale
Examens nécessaires :
Examen de l’acuité visuelle, du strabisme et du fond d’œil : évaluation de la fonction visuelle
Test de traction sous anesthésie générale : vérification de la présence de brides
TDM/IRM : évaluation de la morphologie oculaire et cérébrale. Utile pour confirmer les complications du système nerveux central telles que les anomalies de la gyration occipitale1)
Le panel de tests FRAS/FREM est important pour confirmer le diagnostic 1). En raison du chevauchement des phénotypes du syndrome de Fraser et du syndrome MOTA, il peut être difficile de les distinguer uniquement sur la base des signes cliniques sans test génétique.
Le diagnostic différentiel comprend la colobome palpébral congénital (en cas de déficit partiel), la cyclopie et l’anisocorie.
L’objectif du traitement varie selon le type. Pour les formes complètes et incomplètes, la reconstruction esthétique est l’objectif principal, et le pronostic d’amélioration de la vision est très limité. Dans les formes partielles, la prise en charge du risque de kératopathie d’exposition et de troubles visuels est une priorité.
Gestion de la surface oculaire : Prescription de lubrifiants ophtalmiques et de larmes artificielles (pour lutter contre l’exposition et la sécheresse). En cas de défaut important, utiliser une pommade ophtalmique pour prévenir la sécheresse cornéenne.
Prothèse palpébrale : Option en cas de contre-indication, d’impossibilité ou d’échec de la chirurgie.
Étape 1 : Après incision cutanée du moignon oculaire, insérer un conformateur recouvert d’une greffe muqueuse pour créer le cul-de-sac conjonctival.
Étape 2 (environ 1 an plus tard) : Réaliser une reconstruction palpébrale avec renforcement de la lamelle postérieure et greffe muqueuse orbitaire. En cas d’échec de la greffe muqueuse, le prépuce peut être utilisé comme alternative.
Type incomplet / partiel
Type incomplet : Après création d’un cul-de-sac conjonctival (greffe de muqueuse + mise en place d’un conformateur), une reconstruction palpébrale est réalisée par partage palpébral ou technique de switch. Il existe un risque de récidive de symblépharon cornéo-palpébral.
Type partiel : L’objectif principal est la reconstruction de la paupière supérieure et du fornix supérieur. Une reconstruction en un temps utilisant une greffe de sclère et d’amnios est réalisée.
Si le défaut chez l’enfant est petit, une suture bout à bout peut le réparer. S’il est grand, une chirurgie plastique utilisant un lambeau cutané est choisie. Chez l’enfant rapporté dans le cas clinique, l’exploration oculaire a révélé un globe kystique résiduel et adhérent, et aucune chirurgie supplémentaire n’a été réalisée1).
QComment se déroule la chirurgie du cryptophtalmos complet ?
A
Elle est réalisée par étapes. Lors de la première étape, après incision cutanée du résidu oculaire, un conformateur recouvert d’une greffe muqueuse est inséré pour créer un sac conjonctival. Environ un an plus tard, lors de la deuxième étape, une reconstruction palpébrale avec renforcement de la lamelle postérieure et greffe muqueuse orbitaire est effectuée. L’objectif est principalement l’amélioration esthétique, le pronostic d’amélioration visuelle étant extrêmement rare.
Aucun consensus sur la physiopathologie n’a encore été établi.
Défaut neuroectodermique : La vésicule optique neuroectodermique est essentielle au développement du cristallin fœtal, et un défaut de cette couche empêche le développement approprié de la cornée, du cristallin et de la chambre antérieure.
Trouble de la formation des paupières : Les paupières ne se forment pas sans différenciation de l’ectoderme et du mésoderme. Elles apparaissent à la 6e semaine de gestation et les paupières supérieure et inférieure restent fusionnées jusqu’au 7e mois. Une malformation pendant cette période entraîne des anomalies congénitales des paupières.
Théorie du défaut d’apoptose : En raison de l’association avec la syndactylie, des anomalies laryngées et génitales, on pense qu’un défaut de mort cellulaire programmée joue un rôle important.
Dysfonctionnement du complexe FRAS/FREM : Les mutations de FRAS1, FREM1 et FREM2 entraînent un défaut d’adhésion entre la membrane basale et l’épithélium, provoquant une non-séparation des paupières1).
Les complications secondaires fréquemment associées à la microphtalmie comprennent la cataracte, la luxation du cristallin, le glaucome et le décollement de la rétine.
7. Recherches récentes et perspectives futures (rapports en phase de recherche)
Mwipopo et al. (2023) ont rapporté un cas de cryptophtalmie bilatérale en Tanzanie1). Les tests génétiques ont identifié une mutation FREM2 (hétérozygote probablement pathogène) ainsi qu’une mutation CEP85L (lissencéphalie 10 : LIS10) concomitante. La LIS10 présente une hétérogénéité allant d’un léger retard mental à un phénotype sévère. Ce cas est le premier rapporté en Afrique, et il a été souligné que l’absence de signalement dans les pays à revenu faible ou intermédiaire contribue à une sous-estimation du nombre de cas.
Les mutations des gènes associés au complexe FRAS/FREM présentent des phénotypes chevauchants dans plusieurs syndromes, de sorte que le diagnostic clinique seul est insuffisant et le rôle des tests génétiques s’accroît1). La nécessité des tests génétiques augmente également dans les pays à revenu faible ou intermédiaire1).
Mwipopo E, Massomo MM, Moshiro R, Manji KP. Bilateral cryptophthalmos with overlapping features of Manitoba oculo-tricho-anal (MOTA) syndrome and Fraser syndrome 2. BMJ Case Rep. 2023;16(7):e252618.
Thomas IT, Frias JL, Felix V, Sanchez de Leon L, Hernandez RA, Jones MC. Isolated and syndromic cryptophthalmos. Am J Med Genet. 1986;25(1):85-98. PMID: 3099574.
Kabra M, Gulati S, Ghosh M, Menon PS. Fraser-cryptophthalmos syndrome. Indian J Pediatr. 2000;67(10):775-8. PMID: 11105430.
Copiez le texte de l'article et collez-le dans l'assistant IA de votre choix.
Article copié dans le presse-papiers
Ouvrez un assistant IA ci-dessous et collez le texte copié dans la conversation.