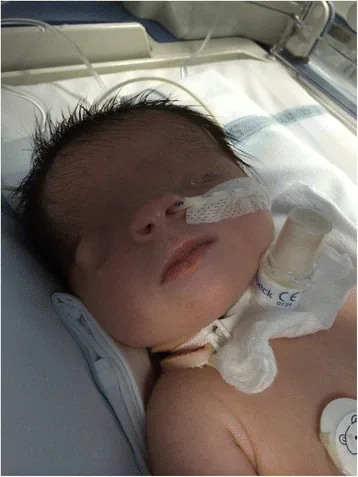
Criptoftalmo deriva dal latino e significa “occhio nascosto”. Si riferisce a una condizione in cui la pelle palpebrale fusa copre il bulbo oculare e l’orbita, con assenza della rima palpebrale. La pelle della fronte è continua con quella della guancia.
L’incidenza è riportata come 0,043 per 10.000 nati vivi e 1,1 per 10.000 nati morti1). Al 2018, solo 55 casi sono stati documentati in letteratura. La prevalenza complessiva delle anomalie palpebrali è dello 0,06%, con 2/3 sporadici e 1/3 con predisposizione genetica1).
Il criptoftalmo è classificato in tre tipi in base alla morfologia.
Tipo completo (tipico): obliterazione completa dell’orbita. È la forma più grave, associata a assenza di sopracciglia, ciglia e strutture ghiandolari.
Tipo incompleto (atipico): residui di palpebre rudimentali. È presente una piccola sacca congiuntivale lateralmente.
Forma abortiva (abortive form / aderenza congenita palpebro-oculare): Assenza della palpebra superiore. La pelle della fronte aderisce alla parte superiore della cornea.
Tutti i tipi possono essere unilaterali o bilaterali, isolati o sindromici. L’occhio nascosto è coperto dalla pelle palpebrale fusa e può presentarsi da solo o come uno dei sintomi della sindrome di Fraser. Se il margine palpebrale non è affatto formato, si diagnostica assenza di palpebre o occhio nascosto.
È strettamente associato alla sindrome di Fraser, spesso dovuta a mutazioni autosomiche recessive nei geni FRAS1, GRIP1 e FREM2. L’80-93% dei pazienti con sindrome di Fraser presenta criptoftalmo.
QUn bambino con criptoftalmo può recuperare la vista?
A
Quasi tutti i casi presentano una funzione visiva scarsa o assente. Lo scopo dell’intervento è principalmente estetico e di ricostruzione dell’area orbitale; il miglioramento della vista è estremamente raro. Per i dettagli, vedere la sezione “Trattamento standard”.
De Bernardo G, Giordano M, Di Toro A, et al. Prenatal diagnosis of Fraser syndrome: a matter of life or death? Ital J Pediatr. 2015 Nov 9;41:86. Figure 1. PMCID: PMC4640198. License: CC BY.
Fotografia del volto subito dopo la nascita: la rima palpebrale non si è formata e la pelle ricopre la superficie oculare. L’occhio sinistro è microftalmico, mentre l’occhio destro presenta anomalie morfologiche; questa immagine aiuta a comprendere l’aspetto del criptoftalmo.
Di seguito sono riportati i principali reperti clinici per tipo.
Tipo completo
Occlusione orbitaria: la pelle della fronte e delle guance è continua e la rima palpebrale è completamente assente.
Assenza degli annessi: sopracciglia, ciglia e strutture ghiandolari sono assenti.
Associazione con microftalmia: la pelle superficiale è fusa con la cornea. Non c’è sacco congiuntivale, la microftalmia è molto comune. Possono essere presenti cisti orbitarie.
Forma incompleta
Forma incompleta: residui di palpebre rudimentali. La lunghezza della rima palpebrale è circa 1/3 del normale. C’è un piccolo sacco congiuntivale lateralmente. L’occhio è piccolo e quasi completamente coperto dalla pelle.
Forma incompleta: assenza della palpebra superiore. La pelle della fronte aderisce al 75% superiore della cornea. La cornea coperta si cheratinizza e diventa opaca, mentre la cornea esposta può rimanere trasparente.
In un caso clinico, un neonato maschio di 39 settimane di gestazione presentava criptoftalmo bilaterale, assenza di sopracciglia, irregolarità dell’attaccatura dei capelli frontali, fessura della punta nasale, orecchie basse, ipertelorismo e malformazione anorettale bassa1). All’occhio destro si palpava una cisti orbitaria mobile di 1×1 cm, mentre il bulbo oculare sinistro era profondamente incassato1). La TC mostrava un pattern anormale di circonvoluzioni cerebrali occipitali e riduzione del bulbo oculare sinistro1).
La causa del criptoftalmo è una mutazione genetica delle proteine che compongono il complesso FRAS/FREM.
Complesso FRAS/FREM: le proteine FRAS1, FREM1 e FREM2 mantengono l’adesione tra membrana basale ed epitelio durante lo sviluppo embrionale. Le mutazioni causano difetti di adesione, portando a anchiloblefaron 1).
Sindrome di Fraser (FRASRS1/2): ereditarietà autosomica recessiva dovuta a mutazioni nei geni FRAS1 o FREM2 1).
Sindrome MOTA: mutazioni di FREM1 associate a fessura nasale, malformazioni anorettali e agenesia renale 1).
Criptoftalmo isolato: le mutazioni di FREM2 possono anche causare criptoftalmo isolato unilaterale o bilaterale 1).
Modalità di trasmissione genetica: prevalentemente autosomica recessiva. Sono stati riportati anche casi di trasmissione autosomica dominante.
I meccanismi noti alla base della coloboma palpebrale congenito includono la mancata fusione delle fessure facciali durante il periodo embrionale e la compressione da parte di bande amniotiche.
QIn che modo la criptoftalmia è correlata alla sindrome di Fraser?
A
La criptoftalmia è presente nell’80-93% dei casi di sindrome di Fraser, causata principalmente da mutazioni nei geni FRAS1 e FREM2. Il complesso FRAS/FREM è essenziale per mantenere l’adesione tra membrana basale ed epitelio durante lo sviluppo embrionale, e la sua mutazione porta a una mancata separazione delle palpebre 1). Tuttavia, le mutazioni di FREM2 possono anche causare criptoftalmia isolata.
È rilevabile intorno alla 18ª settimana di gravidanza tramite ecografia prenatale. I segni sono l’assenza della rima palpebrale e la pelle continua dalla fronte alla guancia. Se sono presenti sindattilia, aumento dell’ecogenicità polmonare e oligoidramnios, è molto probabile la sindrome di Fraser.
I criteri diagnostici per la sindrome di Fraser sono i seguenti.
Classificazione
Elemento
Criterio maggiore
Criptoftalmo, sindattilia, anomalie genitali, anomalie degli arti
Criterio minore
Malformazioni di orecchio e naso, labbro leporino e palatoschisi, anomalie dell’attaccatura dei capelli, malformazioni renali
Esami necessari:
Esame della vista, strabismo e fundus oculi: valutazione della funzione visiva
Test di trazione in anestesia generale: verifica della presenza di bande fibrose
TC/RM: valutazione della morfologia del bulbo oculare e del cervello. Utile per identificare complicanze del sistema nervoso centrale come pattern anomali di circonvoluzione occipitale1)
Il pannello FRAS/FREM è importante per la conferma diagnostica 1). Poiché la sindrome di Fraser e la sindrome MOTA hanno fenotipi sovrapposti, la diagnosi differenziale basata solo sui reperti clinici senza test genetici può essere difficile.
La diagnosi differenziale include coloboma palpebrale congenito (in caso di difetto parziale), ciclopia e anisocoria.
L’obiettivo del trattamento varia a seconda del tipo. Nei tipi completo e incompleto, la ricostruzione estetica è l’obiettivo principale e la prognosi per il miglioramento della vista è estremamente limitata. Nel tipo incompleto, è urgente affrontare il rischio di cheratopatia da esposizione e disturbi visivi.
Gestione della superficie oculare: prescrizione di lubrificanti oftalmici e lacrime artificiali (per contrastare esposizione e secchezza). Se il difetto è ampio, utilizzare unguenti oftalmici per prevenire la secchezza corneale.
Protesi palpebrale: opzione in caso di controindicazione, impossibilità o insuccesso dell’intervento chirurgico.
Primo stadio: dopo l’incisione cutanea del residuo oculare, si inserisce un conformatore rivestito con innesto mucoso per creare il sacco congiuntivale.
Secondo stadio (dopo circa 1 anno): si esegue la ricostruzione palpebrale con rinforzo della lamina posteriore e innesto mucoso orbitario. In caso di fallimento dell’innesto mucoso, si può utilizzare il prepuzio come alternativa.
Tipo incompleto / parziale
Tipo incompleto: dopo la creazione del sacco congiuntivale (innesto di mucosa + posizionamento di conformer), si esegue la ricostruzione palpebrale mediante condivisione palpebrale o tecnica switch. Esiste il rischio di recidiva dell’adesione corneo-palpebrale.
Tipo parziale: l’obiettivo principale è la ricostruzione della palpebra superiore e del fornice superiore. Si esegue una ricostruzione in un unico tempo con innesto di sclera e membrana amniotica.
Nei bambini, se il difetto è piccolo, può essere riparato con sutura diretta. Se è grande, si sceglie un intervento di chirurgia plastica con lembo cutaneo. Nel caso riportato, l’esplorazione oculare ha rivelato un globo oculare cistico residuo e adeso, e non sono stati eseguiti ulteriori interventi1).
QCome viene eseguito l'intervento chirurgico per la criptoftalmia completa?
A
Viene eseguito in fasi. Nella prima fase, dopo l’incisione cutanea del residuo oculare, viene inserito un conformatore rivestito con innesto mucoso per creare un sacco congiuntivale. Circa un anno dopo, nella seconda fase, si esegue la ricostruzione palpebrale con rinforzo della lamina posteriore e innesto mucoso orbitario. L’obiettivo è principalmente il miglioramento dell’aspetto estetico, mentre la prognosi per il miglioramento visivo è estremamente rara.
Non è ancora stato stabilito un consenso unanime sulla fisiopatologia.
Difetto neuroectodermico: la vescicola ottica neuroectodermica è essenziale per lo sviluppo del cristallino fetale, e un difetto in questo strato impedisce il corretto sviluppo di cornea, cristallino e camera anteriore.
Alterazione della formazione palpebrale: le palpebre non si formano senza la differenziazione di ectoderma e mesoderma. Le palpebre compaiono alla sesta settimana di gestazione e le palpebre superiore e inferiore rimangono fuse fino al settimo mese. Un’alterazione in questo processo causa anomalie palpebrali congenite.
Teoria del difetto di apoptosi: la presenza di sindattilia, anomalie laringee e genitali suggerisce che un difetto nella morte cellulare programmata giochi un ruolo importante.
Disfunzione del complesso FRAS/FREM: le mutazioni di FRAS1, FREM1 e FREM2 causano un’alterazione dell’adesione tra membrana basale ed epitelio, portando a mancata separazione palpebrale1).
Le complicanze secondarie che tendono a verificarsi con la microftalmia includono cataratta, lussazione del cristallino, glaucoma e distacco della retina.
7. Ricerche recenti e prospettive future (rapporti in fase di studio)
Mwipopo et al. (2023) hanno riportato un caso di criptoftalmo bilaterale dalla Tanzania1). I test genetici hanno identificato una mutazione FREM2 (eterozigote likely pathogenic) insieme a una mutazione CEP85L (lissencefalia 10: LIS10). La LIS10 mostra eterogeneità, da lieve ritardo mentale a fenotipi gravi. Questo caso è il primo riportato dall’Africa, ed è stato sottolineato che la mancata segnalazione nei paesi a basso e medio reddito contribuisce alla sottostima del numero di casi.
Poiché le mutazioni genetiche correlate al complesso FRAS/FREM mostrano fenotipi sovrapposti in diverse sindromi, la sola diagnosi clinica è insufficiente e il ruolo dei test genetici è in aumento1). La necessità di test genetici sta crescendo anche nei paesi a basso e medio reddito1).
Mwipopo E, Massomo MM, Moshiro R, Manji KP. Bilateral cryptophthalmos with overlapping features of Manitoba oculo-tricho-anal (MOTA) syndrome and Fraser syndrome 2. BMJ Case Rep. 2023;16(7):e252618.
Thomas IT, Frias JL, Felix V, Sanchez de Leon L, Hernandez RA, Jones MC. Isolated and syndromic cryptophthalmos. Am J Med Genet. 1986;25(1):85-98. PMID: 3099574.
Kabra M, Gulati S, Ghosh M, Menon PS. Fraser-cryptophthalmos syndrome. Indian J Pediatr. 2000;67(10):775-8. PMID: 11105430.
Copia il testo dell'articolo e incollalo nell'assistente IA che preferisci.
Articolo copiato negli appunti
Apri un assistente IA qui sotto e incolla il testo copiato nella chat.