آتاکسی فریدریش (FRDA) یک بیماری پیشرونده ژنتیکی است که سیستم عصبی مرکزی و محیطی را درگیر میکند. این بیماری شایعترین آتاکسی ارثی با توارث اتوزومال مغلوب در جمعیت سفیدپوستان محسوب میشود.
در سال ۱۸۶۳، نیکولاس فریدریش آتاکسی با شروع جوانی، اسکولیوز و کاردیومیوپاتی خانوادگی را گزارش کرد. بعدها، پیر ماری FRDA را از سایر آتاکسیها متمایز کرد و مفهوم این بیماری تثبیت شد.
از نظر اپیدمیولوژیک، تفاوتهای منطقهای زیادی وجود دارد و شیوع آن از ۱:۲۰,۰۰۰ تا ۱:۷۵۰,۰۰۰ گزارش شده است. در اروپاییها حدود ۱:۲۱,۰۰۰1) و در سراسر جهان ۱:۴۰,۰۰۰2) تا ۱:۵۰,۰۰۰3) تخمین زده میشود. فراوانی ناقلان حدود ۱/۷۰ است1). شیوع در جنوب فرانسه، شمال اسپانیا و ایرلند بالا و در اسکاندیناوی و روسیه پایین است. در جنوب صحرای آفریقا و نیمکره شرقی حتی کمتر است، اما موارد تأیید شده ژنتیکی در غرب آفریقا (خانوادههای توآرگ با ازدواج فامیلی در مالی) نیز گزارش شده است3).
الگوی توارث اتوزومال مغلوب است و میزان بروز در مردان و زنان برابر است. میانگین سن شروع علائم ۱۵.۵ سال است و بیشتر موارد قبل از ۲۵ سالگی شروع میشود. شروع در سنین ۸ تا ۱۵ سال شایعتر است2). میانگین امید به زندگی ۳۹ سال است و علت اصلی مرگ کاردیومیوپاتی است2).
Qآتاکسی فریدریش با چه فراوانی رخ میدهد؟
A
شایعترین آتاکسی ارثی در جمعیت سفیدپوستان است و شیوع آن بسته به منطقه جغرافیایی از ۱ در ۲۰٬۰۰۰ تا ۱ در ۷۵۰٬۰۰۰ متغیر است. در اروپا حدود ۱ در ۲۱٬۰۰۰ نفر1) و در سراسر جهان ۱ در ۴۰٬۰۰۰ تا ۵۰٬۰۰۰ نفر تخمین زده میشود2)3). فراوانی ناقلان حدود ۱ در ۷۰ گزارش شده است1).
Petya Bogdanova-Mihaylova, Helena Maria Plapp, Hongying Chen et al. Longitudinal Assessment Using Optical Coherence Tomography in Patients with Friedreich’s Ataxia. Tomography. 2021 Dec 8; 7(4):915. Figure 1. PMCID: PMC8706975. License: CC BY.
اختلال راه رفتن: شایعترین علامت اولیه. با راه رفتن ناپایدار در همه جهات شروع میشود.
ناهماهنگی و لرزش اندام فوقانی: در برخی موارد به صورت لرزش دست شروع میشود3).
دیسآرتری (اختلال تکلم): اغلب در عرض ۱۰ تا ۱۵ سال پس از شروع بیماری ظاهر میشود.
علائم حسی: از دست دادن حس عمقی (حس عمقی) رخ میدهد.
علائم بینایی: در برخی بیماران کاهش بینایی و کاهش حساسیت کنتراست رخ میدهد، اما اکثر بیماران در مراحل اولیه علائم بینایی ندارند.
پیشرفت علائم حرکتی در افراد متفاوت است، اما به طور متوسط حدود ۸ سال پس از اولین علائم، بیمار قادر به راه رفتن مستقل نیست و در ۱۱ تا ۱۵ سال به ویلچر نیاز پیدا میکند. در مواردی با تعداد تکرار GAA بالا، گزارش شده که در ۵ سال به ویلچر نیاز پیدا کردهاند3).
یافتههای بالینی (یافتههایی که پزشک در معاینه تأیید میکند)
راه رفتن آتاکسیک: راه رفتن ناپایدار در همه جهات. همراه با دیسمتری، هیپوتونی، دیسدیادوکوکینزی و اختلال در زمان هماهنگی حرکات1).
عدم وجود رفلکسهای عمقی تاندون: عدم وجود رفلکس کشکک و آشیل جزء معیارهای تشخیصی هاردینگ است. عدم وجود رفلکس اندام فوقانی نیز شایع است. در برخی موارد، هیپررفلکسی، اسپاستیسیته و راه رفتن قیچیوار دیده میشود3).
علامت بابینسکی: به صورت پاسخ کف پایی اکستانسور ظاهر میشود.
عوارض سیستمیک: کاردیومیوپاتی هیپرتروفیک (تا ۶۳٪)، دیابت (۵ تا ۴۰٪)2).
یافتههای نورو-افتالمولوژیک
نیستاگموس (nystagmus): ناشی از اختلال در مدارهای مخچه-ساقه مغز. شامل نیستاگموس وابسته به وضعیت چشم.
حرکات جهشی مربعی (square-wave jerks): حرکات ساکادیک غیرارادی که در حین تثبیت نگاه رخ میدهد.
اختلال حرکات تعقیبی: ردیابی هدف به صورت پلهای (non-smooth pursuit).
دیسمتری ساکادیک (saccadic dysmetria): ساکادهای بیش از حد یا کمتر از حد.
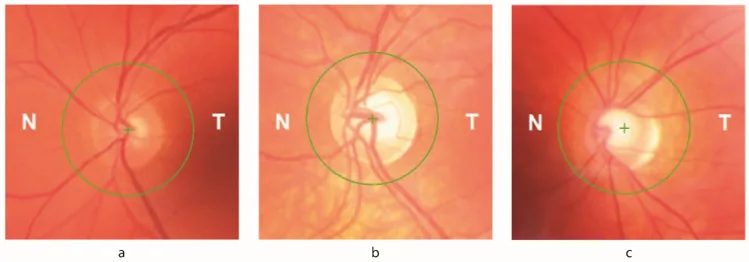
آتروفی عصب بینایی: در معاینه فوندوس مشاهده میشود. تا ۳۰٪ از بیماران علائم چشمی دارند.
کاهش ضخامت لایه فیبرهای عصبی شبکیه (RNFL) : با OCT قابل تشخیص است. با کاهش حدت بینایی و حساسیت کنتراست ارتباط مستقیم دارد.
نقص میدان بینایی : نقص اولیه میدان بینایی که از محیط به صورت دایرهای پیشرفت میکند. با از دست رفتن RNFL ثبت شده در OCT مرتبط است.
Qآیا علائم چشمی همیشه بروز میکنند؟
A
در FRDA، تا ۳۰٪ از بیماران علائم چشمی دارند، اما بسیاری از بیماران در مراحل اولیه علائم بینایی ذهنی ندارند. آتروفی عصب بینایی، کاهش ضخامت RNFL و اختلال حرکات چشم با معاینات عینی قابل تشخیص هستند و با کاهش حدت بینایی و حساسیت کنتراست ارتباط مستقیم دارند. معاینات منظم چشم پزشکی توصیه میشود.
علت FRDA افزایش تکرار سهنوکلئوتیدی GAA در اینترون ۱ ژن FXN (فراتاکسین) روی کروموزوم ۹ است. ۹۶٪ موارد هموزیگوت برای افزایش پاتوژنیک دو آللی هستند1). موارد باقیمانده ترکیبی از افزایش GAA و جهش نقطهای، یا افزایش GAA و حذف درونژنی/کل ژن به صورت هتروزیگوت مرکب هستند1).
رابطه بین تعداد تکرار GAA و بیماری در زیر نشان داده شده است.
طبقهبندی
تعداد تکرار GAA
طبیعی
۵ تا ۳۳ بار
میانی (معادل ناقل)
۳۴ تا ۶۵ بار
بیماریزا
۶۶ بار یا بیشتر (در برخی گزارشها ۹۰ بار یا بیشتر)1)3)
هرچه تعداد تکرارهای GAA بیشتر باشد، سن شروع بیماری زودتر و شدت آن بیشتر است. در یک مورد با تعداد تکرارهای آللی بسیار طولانی 999/766، بیماری در سن ۱۱ سالگی شروع شد و در عرض ۵ سال نیاز به ویلچر پیدا کرد (سریعتر از میانگین ۱۰ ساله) 3). وقفه (interruption) درون توالی تکراری ممکن است سن شروع بیماری را به تأخیر بیندازد 1).
هتروزیگوتهای مرکب از حذف درونژنی (intragenic deletion) و افزایش تکرار GAA نسبت به موارد افزایش تکرار دو آللی، تمایل به شروع زودتر، پیشرفت سریعتر و کاردیومیوپاتی شدیدتر دارند، اگرچه مواردی با سیر بالینی معمولی نیز گزارش شده است 1).
ازدواج فامیلی به دلیل ماهیت اتوزومال مغلوب بیماری، خطر ابتلا را افزایش میدهد 2)3). این بیماری در جمعیتهای با نیای اروپایی شیوع بیشتری دارد.
Qاگر آزمایش ژنتیک نشاندهنده هموزیگوت بودن باشد، آیا احتمال هتروزیگوت مرکب وجود دارد؟
A
تجزیه و تحلیل قطعات PCR و TP-PCR قادر به تشخیص حذف درون ژنی نیستند، بنابراین گسترش دو آللی ظاهری ممکن است در واقع ترکیبی از گسترش GAA و حذف درون ژنی باشد 1). آزمایش تکمیلی با MLPA (تقویت وابسته به اتصال پروب چندگانه) و بررسی نمونههای والدین برای مشاوره ژنتیکی دقیق ضروری است.
PCR + TP-PCR: تکرار GAA در اینترون 1 را تکثیر کرده و آلل طویل شده را تشخیص میدهد.
MLPA (Multiplex Ligation-dependent Probe Amplification): برای تشخیص حذفها و تکراریهایی که با آنالیز قطعه یا TP-PCR قابل تشخیص نیستند، ضروری است1). برای رد احتمال هتروزیگوت مرکب در مواردی که به نظر میرسد گسترش دو آللی وجود دارد، اضافه میشود.
آنالیز ساترن بلات: در ترکیب با PCR، تشخیص با دقت بیش از 99% امکانپذیر است3).
بررسی نمونه والدین: برای مشاوره ژنتیکی دقیق ضروری است1).
الکترواکولوگرافی (EOG): برای تحلیل کیفی و کمی حرکات چشم استفاده میشود. ساکاد هدایتشده بینایی (تأخیر و دامنه)، تعقیب (بهره در سرعتهای مختلف هدف)، VOR (بهره در سرعتهای مختلف سر)، و تثبیت (تحلیل شکل موج نیستاگموس، تأخیر موج مربعی) ارزیابی میشود.
توموگرافی انسجام نوری (OCT): ضخامت لایه فیبرهای عصبی شبکیه (RNFL) را به صورت کمی اندازهگیری میکند. همبستگی با کاهش حدت بینایی و حساسیت کنتراست قابل تأیید است.
بررسی میدان بینایی: الگوی نقص پیشرونده به صورت دایرهای از محیط به مرکز ارزیابی میشود.
بررسی فوندوس: وجود یا عدم وجود آتروفی عصب بینایی تأیید میشود.
نوار قلب و اکوکاردیوگرافی : برای ارزیابی کاردیومیوپاتی، ضخامت دیواره بطن چپ، کسر جهشی و عملکرد دیاستولیک 2). پایش دورهای ضروری است.
آزمایشهای مرتبط با قند خون2): قند خون ناشتا، HbA1c، پپتید C، انسولین ناشتا، آنتیبادیهای خودایمنی جزایر پانکراس (GAD65، IA-2، ZnT8)، HOMA2-IR و HOMA2-%B برای تشخیص زودهنگام دیابت و تعیین نوع آن انجام میشود.
مقیاسهای ارزیابی بالینی: FARS (مقیاس رتبهبندی آتاکسی فریدریش)، ICARS و SARA برای ارزیابی کمی علائم عصبی استفاده میشود2). با SARA، بدتر شدن سالانه 0.77 امتیاز (خطای استاندارد 0.06) در سیر طبیعی گزارش شده است2).
MRI مغز و نخاع: آتروفی مخچه (در برخی موارد 3 سال پس از شروع بیماری ظاهر میشود3)) و تغییرات دژنراتیو نخاع ارزیابی میشود.
مدیریت اسکولیوز: برای موارد خفیف تا متوسط از بریس و برای موارد شدید جراحی در نظر گرفته میشود.
مدیریت پای توخالی: تزریق سم بوتولینوم به عضله گاستروکنمیوس و کشش تاندون آشیل برای بهبود تحرک.
توانبخشی: حفظ عملکرد از طریق فیزیوتراپی، کاردرمانی و گفتاردرمانی.
چشمپزشکی
مراقبت از کمبینایی: استفاده از ذرهبین، تنظیم نور و آموزش زندگی روزمره برای بیماران با علائم بینایی ناشی از آتروفی عصب بینایی و آتروفی شبکیه.
درمان علامتی اختلالات حرکات چشم: برای نیستاگموس وابسته به وضعیت چشم از عینک منشوری (افزودن درجه منشور یکسان به هر دو چشم در جهت بدترکننده وضعیت) استفاده میشود. برای نیستاگموس عمودی، نیستاگموس متناوب دورهای و حرکات ساکادیک، داروهای آگونیست گیرنده GABA_B تجویز میشود.
مثال نسخه (آگونیست گیرنده GABA_B): قرص گابارون (5 میلیگرم) 3 تا 6 قرص، یک تا سه بار در روز.
دیابت همراه با FRDA به دلیل اختلال عملکرد میتوکندریایی زمینهای، نیاز به احتیاط در انتخاب دارو دارد2).
داروهایی که باید اجتناب کرد: متفورمین و تیازولیدیندیونها به دلیل مهار کمپلکس I میتوکندری باید اجتناب شوند2). سولفونیلاورهها خطر استرس سلولهای بتا و هیپوگلیسمی را دارند.
داروهای کاهنده قند خون توصیه شده: مهارکنندههای DPP-4 (مانند سیتاگلیپتین 100 میلیگرم در روز) و آنالوگهای GLP-1 ترجیح داده میشوند2). ایمگلیمین (500 میلیگرم دو بار در روز) ممکن است به عنوان یک داروی جدید کاهنده قند خون با هدف قرار دادن عملکرد میتوکندری مفید باشد2).
انسولین درمانی: با دوز 0.5 واحد به ازای هر کیلوگرم وزن بدن در روز شروع کنید2).
درمان کمکی هدفمند میتوکندری: ترکیب ال-کارنیتین 500 میلیگرم در روز، کوآنزیم Q10 100 میلیگرم در روز و ویتامین E 400 واحد بینالمللی در روز گزارش شده است2).
در یک گزارش موردی با استفاده از رژیم فوق، HbA1c از 13.3% به 8.4% (پس از 17 ماه) و سپس به 6.9% (پس از 19 ماه اضافی) بهبود یافت و ICARS از 85 به 71 کاهش یافت (14 امتیاز بهبود)2).
Qچه درمانی برای دیابت همراه با آتاکسی فریدریش مناسب است؟
A
از آنجا که اختلال عملکرد میتوکندری زمینهای وجود دارد، از مصرف متفورمین و تیازولیدیندیونها خودداری میشود 2). مهارکنندههای DPP-4 (مانند سیتاگلیپتین) و آنالوگهای GLP-1 توصیه میشوند. درمانهای کمکی هدفمند میتوکندری مانند ال-کارنیتین، CoQ10 و ویتامین E نیز ممکن است مفید باشند 2).
فراتاکسین پروتئینی است که در غشای داخلی میتوکندری قرار دارد. در متابولیسم آهن (ذخیره آهن و مونتاژ خوشههای آهن-گوگرد) نقش داشته و برای عملکرد طبیعی زنجیره تنفسی میتوکندری ضروری است 1).
افزایش توالی تکراری GAA باعث خاموشی رونویسی (transcriptional silencing) شده و سطح mRNA FXN را کاهش میدهد 1). کمبود فراتاکسین منجر به موارد زیر میشود.
تجمع آهن در میتوکندری: به دلیل کاهش عملکرد پردازش آهن.
کاهش تولید ATP: به دلیل اختلال عملکرد کمپلکس زنجیره تنفسی2).
کمبود فراتاکسین باعث تغییر سطح آهن داخل سلولی شده و سلولهای گانگلیونی شبکیه (RGC) را در برابر استرس اکسیداتیو آسیبپذیر میکند. تا ۳۰٪ از بیماران FA علائم چشمی نشان میدهند. هر دو مدار ساقه مغز-مخچه و عصب بینایی تحت تأثیر قرار میگیرند. کاهش ضخامت RNFL با کاهش حدت بینایی و حساسیت کنتراست همبستگی مستقیم دارد و نقص اولیه میدان بینایی که به صورت متحدالمرکز از محیط پیشرفت میکند، با از دست دادن RNFL ثبت شده در OCT مرتبط است.
اختلال عملکرد میتوکندری علت اصلی دیابت مرتبط با FRDA است2).
اختلال عملکرد سلولهای بتا: کاهش ترشح انسولین به دلیل اختلال در تولید ATP و در نهایت از بین رفتن سلولهای بتا.
تنظیم غیرطبیعی ترشح سلولهای آلفا: هیپرگلوکاگونمی متناقض در هیپرگلیسمی و پاسخ ناکافی گلوکاگون در هیپوگلیسمی2).
مکانیسم مقاومت به انسولین: کاهش جذب گلوکز به دلیل اختلال در فسفوریلاسیون اکسیداتیو عضلات اسکلتی، تجمع چربی نابجا در کبد و عضله به دلیل اختلال متابولیسم لیپید، اختلال در سیگنالدهی انسولین به دلیل التهاب مزمن و استرس اکسیداتیو، تولید غیرطبیعی گلوکز کبدی به دلیل نوروپاتی اتونوم، و بدتر شدن ثانویه متابولیسم به دلیل عدم فعالیت بدنی و آتروفی عضلانی ناشی از آتاکسی2).
در افراد دارای حذف درونژنی (Intragenic deletion)، تمایل به شروع زودتر، پیشرفت سریعتر و کاردیومیوپاتی شدیدتر نسبت به افراد با گسترش دو آللی (biallelic expansion) وجود دارد1). اگر حذف باعث حذف کدون شروع شود، تولید پروتئین به طور کامل از بین میرود1).
Qچرا در آتاکسی فریدریش علائم چشمی ایجاد میشود؟
A
اختلال متابولیسم آهن و استرس اکسیداتیو ناشی از کمبود فراتاکسین به سلولهای گانگلیونی شبکیه آسیب میزند. کاهش ضخامت لایه فیبرهای عصبی شبکیه (RNFL) به طور مستقیم با کاهش حدت بینایی و حساسیت کنتراست مرتبط است و باعث نقص میدان بینایی میشود که از محیط به صورت دایرهای پیشرفت میکند. همچنین آسیب به مدارهای ساقه مغز و مخچه باعث ناهنجاریهای حرکات چشم مانند نیستاگموس، اختلال حرکات تعقیبی و اختلال ساکاد میشود.
7. تحقیقات جدید و چشمانداز آینده (گزارشهای در مرحله تحقیق)
کارآزمایی بالینی Cooper et al. (2008) گزارش کرد که درمان ترکیبی CoQ10 و ویتامین E به طور معناداری نمره ICARS را طی دو سال بهبود بخشید2).
در یک کارآزمایی متقاطع تصادفی شده با دارونما، Schöls et al. (2005) نشان داد که تجویز L-کارنیتین تولید ATP میتوکندری را به طور معناداری بهبود میبخشد2).
Sureshkumar و همکاران (2025) یک زن 32 ساله مبتلا به دیابت مرتبط با FA را با ترکیبی از انسولین، سیتاگلیپتین، L-کارنیتین، CoQ10، ویتامین E، ویتامینهای نوروتروپیک و ایمگلیمین درمان کردند2). HbA1c از 13.3% پس از 17 ماه به 8.4% و پس از 19 ماه به 6.9% بهبود یافت و ICARS از 85 به 71 کاهش یافت (14 امتیاز بهبود) (HOMA2-IR: 4.5→1.2، HOMA2-%B: 5→60، MAGE: 120→70 mg/dL، CV: 43%→34.9%). در حالی که تاریخچه طبیعی SARA پیشبینی میکند که طی 3 سال حدود 2.31 امتیاز بدتر شود، این بیمار انحراف مثبت حدود 16 امتیاز نشان داد. این اولین مورد گزارش شده از دستیابی همزمان به تثبیت طولانیمدت قند خون و بهبود آتاکسی است.
تصور میشود ایمگلیمین از طریق بهبود فعالیت زنجیره تنفسی، کاهش استرس اکسیداتیو و افزایش سنتز ATP/NAD+ عمل میکند2).
درمان با ناقل AAV : کارآزمایی بالینی AAVrh.10hFXN با هدف انتقال ژن طبیعی FXN به قلب و سیستم عصبی در حال انجام است و بهبود اولیه در بیان FXN و نشانگرهای بیماری نشان داده شده است (Munoz-Zuluaga et al. 2023)2).
CRISPR-Cas9: با هدف حذف توالیهای تکراری GAA، در سلولهای مشتق از YG8R و مدلهای موشی در حال بررسی است (Ouellet et al. 2017)2).
چالش: کنترل پاسخ ایمنی و ایجاد روش ایمن برای انتقال ژن ضروری است2).
Aguilera و همکاران (2023) با بررسی نمونههای والدین بیمارانی که تصور میشد دارای گسترش دو آللی (biallelic) هستند، یک حذف درونژنی جدید شامل ناحیه 5’UTR و اگزونهای 1 تا 2 از ژن FXN را شناسایی کردند1). تاکنون تنها 10 مورد حذف درونژنی در مقالات گزارش شده است، اما احتمال میرود که فراوانی آن در واقع بیشتر باشد. استانداردسازی آنالیز ژنتیکی تکمیلی با استفاده از روشهایی مانند MLPA و بررسی نمونههای والدین به عنوان چالشهای آینده مطرح شده است.
گزارش اولین مورد تأیید شده در غرب آفریقا نشاندهنده نیاز به مطالعات کوهورت بزرگ در زمینههای قومی و جغرافیایی متنوع است3). شناسایی واریانتهای تعدیلکننده بیماری میتواند در آینده به عنوان اهداف درمانی مورد استفاده قرار گیرد.
Aguilera C, Esteve-Garcia A, Casasnovas C, et al. Novel intragenic deletion within the FXN gene in a patient with typical phenotype of Friedreich ataxia: may be more prevalent than we think? BMC Med Genomics. 2023;16:312.
Sureshkumar P, Kumar SS, Cheriyan J, Masood A. Friedreich Ataxia and Related Diabetes: Therapeutic Approach Targeting Mitochondrial Dysfunction. JCEM Case Rep. 2025;3:luaf215.
Cissé CAK, Cissé L, Ba HO, et al. Friedreich ataxia in a family from Mali, West Africa. Clin Case Rep. 2021;9:e04065.
متن مقاله را کپی کنید و در دستیار هوش مصنوعی دلخواه خود بچسبانید.
مقاله در کلیپبورد کپی شد
یکی از دستیارهای هوش مصنوعی زیر را باز کنید و متن کپیشده را در کادر گفتگو بچسبانید.