رنح فريدريش (Friedreich Ataxia; FRDA) هو مرض وراثي تقدمي يصيب الجهاز العصبي المركزي والمحيطي. يُعتبر أكثر أنواع الرنح الوراثي المتنحي شيوعًا بين السكان البيض.
في عام 1863، وصف نيكولاوس فريدريش حالات من الرنح المبكر، والجنف، واعتلال القلب العائلي. لاحقًا، ميز بيير ماري FRDA عن أنواع الرنح الأخرى، مما أسس المفهوم المرضي.
وبائيًا، هناك تباين جغرافي كبير، حيث تتراوح نسبة الانتشار بين 1:20,000 و1:750,000. بين الأوروبيين، يُقدر بحوالي 1:21,0001)، وعلى مستوى العالم بين 1:40,0002) و1:50,0003). يُقدر تواتر حاملي المرض بحوالي 1/701). تزداد النسبة في جنوب فرنسا وشمال إسبانيا وأيرلندا، وتقل في إسكندنافيا وروسيا. في أفريقيا جنوب الصحراء ونصف الكرة الشرقي، تكون النسبة أقل، ولكن تم تأكيد حالات وراثية في غرب أفريقيا (عائلة طوارق في مالي مع زواج الأقارب)3).
نمط الوراثة هو جسمي متنحي، ومعدل الإصابة متساوٍ بين الذكور والإناث. متوسط عمر ظهور الأعراض هو 15.5 سنة، ومعظم الحالات تظهر قبل سن 25. غالبًا ما يبدأ بين 8 و15 سنة2). متوسط العمر المتوقع هو 39 سنة، والسبب الرئيسي للوفاة هو اعتلال عضلة القلب2).
Qما مدى شيوع رنح فريدريش؟
A
هو أكثر أنواع الرنح الوراثي شيوعًا بين السكان البيض، ويتراوح معدل انتشاره حسب المنطقة من 1:20,000 إلى 1:750,000. في أوروبا، يُقدر بحوالي 1 من كل 21,000 شخص1)، وعلى مستوى العالم بحوالي 1 من كل 40,000 إلى 50,000 شخص2)3). ويُقدر تواتر حاملي المرض بحوالي 1 من كل 70 شخصًا1).
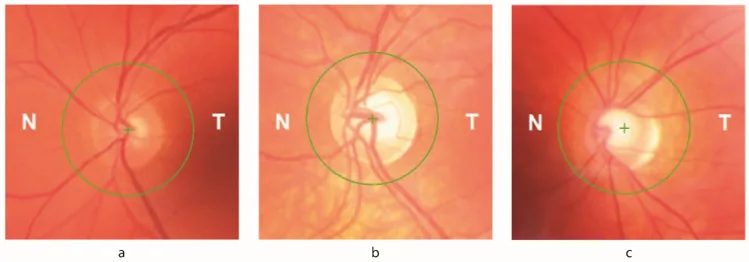
Petya Bogdanova-Mihaylova, Helena Maria Plapp, Hongying Chen et al. Longitudinal Assessment Using Optical Coherence Tomography in Patients with Friedreich’s Ataxia. Tomography. 2021 Dec 8; 7(4):915. Figure 1. PMCID: PMC8706975. License: CC BY.
الأعراض البصرية: قد يعاني بعض المرضى من انخفاض حدة البصر وحساسية التباين، لكن معظمهم لا تظهر لديهم أعراض بصرية في المراحل المبكرة.
يختلف تطور الأعراض الحركية بين الأفراد، لكن في المتوسط، يفقد المريض القدرة على المشي بعد حوالي 8 سنوات من ظهور الأعراض الأولى، ويحتاج إلى كرسي متحرك بعد 11-15 سنة. في حالات تكرار GAA الطويل، تم الإبلاغ عن الحاجة إلى كرسي متحرك خلال 5 سنوات 3).
المشي الرنحي: مشية غير مستقرة في جميع الاتجاهات. مصحوبة بخلل القياس، نقص التوتر، عدم القدرة على الحركات المتناوبة السريعة، واضطراب التوقيت الحركي المشترك 1).
فقدان المنعكسات الوترية العميقة: فقدان منعكس الرضفة ومنعكس وتر أخيل هو عنصر إلزامي في معايير هاردينغ التشخيصية. غالبًا ما يصاحبه فقدان المنعكسات في الأطراف العلوية. قد تظهر حالات مع فرط المنعكسات، التشنج، والمشي المقصي 3).
الرأرأة (nystagmus): بسبب خلل في دوائر المخيخ وجذع الدماغ. تشمل الرأرأة المعتمدة على وضعية العين.
الرمشات المربعة (square-wave jerks): حركات عينية اندفاعية لا إرادية تتداخل أثناء التثبيت.
اضطراب حركات التتبع: يصبح تتبع الهدف متدرجًا (خلل في التتبع البطيء).
خلل قياس الحركات الرمية (saccadic dysmetria): ظهور حركات رمية مفرطة أو ناقصة.
ضمور العصب البصري: يُلاحظ في فحص قاع العين. تظهر علامات عينية لدى 30% من المرضى.
انخفاض سمك طبقة الألياف العصبية الشبكية (RNFL): يُكتشف بواسطة التصوير المقطعي التوافقي البصري (OCT). يرتبط ارتباطًا مباشرًا بانخفاض حدة البصر وحساسية التباين.
عيب المجال البصري: عيوب مبكرة في المجال البصري تتقدم بشكل متحد المركز من المحيط. مرتبطة بفقدان طبقة الألياف العصبية الشبكية المسجل بواسطة التصوير المقطعي التوافقي البصري.
Qهل تظهر الأعراض العينية بالضرورة؟
A
في مرض فريدريك الرنح (FRDA)، تظهر العلامات العينية لدى ما يصل إلى 30% من المرضى، لكن معظم المرضى يفتقرون إلى الأعراض البصرية الذاتية في المراحل المبكرة. يُكتشف ضمور العصب البصري، وانخفاض سمك طبقة الألياف العصبية الشبكية، واضطرابات حركة العين من خلال الفحوصات الموضوعية، وترتبط ارتباطًا مباشرًا بانخفاض حدة البصر وحساسية التباين. يُوصى بإجراء فحوصات عينية دورية.
سبب مرض فريدريك الرنح هو تمدد تكرار ثلاثي النوكليوتيدات GAA في إنترون 1 من جين FXN (فراتاكسين) على الكروموسوم 9. 96% من الحالات تكون متماثلة الزيجوت لتمددات ممرضة ثنائية الأليل1). الحالات المتبقية تكون متغايرة الزيجوت المركبة لتمدد GAA وطفرة نقطية، أو تمدد GAA وحذف داخل الجين/كامل الجين1).
العلاقة بين عدد تكرارات GAA والمرض موضحة أدناه.
التصنيف
عدد تكرارات GAA
طبيعي
5–33 تكرارًا
متوسط (ما يعادل الناقل)
34–65 تكرارًا
مرضي
66 تكرارًا أو أكثر (في تقارير أخرى 90 تكرارًا أو أكثر)1)3)
كلما زاد عدد تكرارات GAA، كان سن البداية أبكر وكانت الحالة أشد. في حالة ذات أليل طويل جدًا (999/766)، بدأ المرض في سن 11 عامًا وأصبح الكرسي المتحرك ضروريًا خلال 5 سنوات (أسرع من المتوسط البالغ 10 سنوات) 3). قد يؤدي الانقطاع داخل التسلسل المتكرر إلى تأخير سن البداية 1).
يُعتقد أن متغاير الزيجوت المركب (حذف داخل الجين + توسع GAA) يؤدي إلى بداية أبكر وتقدم أسرع واعتلال عضلة قلبية أشد مقارنة بالتوسع ثنائي الأليل، ولكن تم الإبلاغ عن حالات ذات مسار نموذجي 1).
لا يمكن لتحليل أجزاء PCR وTP-PCR اكتشاف الحذف داخل الجين، لذلك قد يكون التوسع ثنائي الأليل الظاهر في الواقع توسع GAA + حذف داخل الجين 1). الاختبار الإضافي باستخدام MLPA (تضخيم التابع للربط متعدد المواقع) وفحص عينات الوالدين ضروريان للاستشارة الوراثية الدقيقة.
بداية المرض قبل سن 25 عامًا، ترنح تدريجي في المشي والأطراف، فقدان منعكس الرضفة ووتر العرقوب، دليل على تنكس محوري، عسر الكلام (بعد 5 سنوات من البداية)
النتائج الإضافية (أكثر من 66%)
الجنف، ضعف العضلات القشرية النخاعية في الأطراف السفلية، فقدان المنعكسات في الأطراف العلوية، فقدان الإحساس في الألياف العصبية السميكة، تخطيط كهربائي غير طبيعي للقلب
PCR + TP-PCR: تضخيم تكرار GAA في الإنترون 1 للكشف عن الأليل المطول.
MLPA (تضخيم المسبار المعتمد على الربط المتعدد): ضروري لتشخيص الحذف أو الازدواجية التي لا يمكن اكتشافها بواسطة تحليل الشظايا أو TP-PCR1). يُضاف لاستبعاد احتمال أن يكون التوسع ثنائي الأليل الظاهر متغاير الزيجوت المركب.
تحليل Southern blot: يمكن أن يحقق التشخيص بدقة تزيد عن 99% عند دمجه مع PCR3).
فحوصات السكر ذات الصلة2): يتم إجراء قياس سكر الدم الصائم، HbA1c، الببتيد C، الأنسولين الصائم، الأجسام المضادة الذاتية لجزر البنكرياس (GAD65، IA-2، ZnT8)، HOMA2-IR، HOMA2-%B للكشف المبكر عن مرض السكري وتحديد نوعه.
مقاييس التقييم السريري: يتم التقييم الكمي للأعراض العصبية باستخدام FARS (مقياس تقييم ترنح فريدريش)، ICARS، وSARA2). يُبلغ عن تدهور سنوي بمقدار 0.77 نقطة (خطأ معياري 0.06) في SARA في المسار الطبيعي للمرض2).
التصوير بالرنين المغناطيسي للدماغ والحبل الشوكي: تقييم ضمور المخيخ (مع وجود حالات ظهرت بعد 3 سنوات من ظهور المرض3)) وعلامات تنكس الحبل الشوكي.
إدارة الجنف: في الحالات الخفيفة إلى المتوسطة، يتم استخدام الدعامات. وفي الحالات الشديدة، يُنظر في الجراحة.
إدارة القدم المقوسة: تحسين الحركة عن طريق حقن توكسين البوتولينوم في عضلة الساق وتمديد وتر أخيل.
إعادة التأهيل: الحفاظ على الوظائف من خلال العلاج الطبيعي والوظيفي وعلاج النطق.
طب العيون
رعاية ضعف البصر: استخدام المكبرات وتعديل الإضاءة وتقديم إرشادات الحياة للمرضى الذين يعانون من أعراض بصرية بسبب ضمور العصب البصري أو ضمور الشبكية.
العلاج العرضي لاضطرابات حركة العين: استخدام النظارات المنشورية لرأرأة تعتمد على وضع العين (إضافة قوة منشورية متساوية لكلتا العينين في اتجاه الوضع المتفاقم). إعطاء ناهضات مستقبلات GABA_B لرأرأة عمودية، رأرأة دورية متغيرة الاتجاه، أو الحركات العينية الرمعية المختلطة.
وصفة مثال (ناهض GABA_B): أقراص جابالون (5 ملغ) 3-6 أقراص، مقسمة على 1-3 جرعات.
نظرًا لأن مرض السكري المرتبط بـ FRDA ناتج عن خلل في وظيفة الميتوكوندريا، يجب توخي الحذر عند اختيار الأدوية 2).
الأدوية التي يجب تجنبها: يجب تجنب الميتفورمين والثيازوليدينديون لأنهما يثبطان المركب الأول للميتوكوندريا 2). السلفونيل يوريا يسبب إجهاد خلايا بيتا وخطر نقص سكر الدم.
الأدوية الخافضة لسكر الدم الموصى بها: يفضل استخدام مثبطات DPP-4 (مثل سيتاجليبتين 100 ملغ/يوم) ونظائر GLP-1 2). قد يكون إيميغليمين (500 ملغ مرتين يومياً) مفيداً كدواء جديد لخفض سكر الدم يستهدف وظيفة الميتوكوندريا 2).
العلاج بالأنسولين: يبدأ بجرعة 0.5 وحدة/كغ/يوم كمرجع 2).
العلاج المساعد المستهدف للميتوكوندريا: تم الإبلاغ عن استخدام مزيج من L-كارنيتين 500 ملغ/يوم، CoQ10 100 ملغ/يوم، وفيتامين E 400 وحدة دولية/يوم 2).
في تقرير حالة باستخدام النظام أعلاه، تحسن HbA1c من 13.3% إلى 8.4% (بعد 17 شهراً)، ثم إلى 6.9% (بعد 19 شهراً إضافياً)، وتحسنت درجة ICARS من 85 إلى 71 بفارق 14 نقطة 2).
Qما هو العلاج المناسب لداء السكري المصاحب لمرض فريدريش؟
A
نظرًا لأن الخلل الوظيفي للميتوكوندريا هو الأساس، يجب تجنب الميتفورمين والثيازوليدينديونات 2). يُوصى باستخدام مثبطات DPP-4 (مثل سيتاجليبتين) ونظائر GLP-1. قد تكون العلاجات المساعدة المستهدفة للميتوكوندريا مثل L-كارنيتين وCoQ10 وفيتامين E مفيدة أيضًا 2).
الفرتاكسين هو بروتين موضعي في الغشاء الداخلي للميتوكوندريا. يشارك في استقلاب الحديد (تخزين الحديد وتجميع كبريتيد الحديد) وهو ضروري للوظيفة الطبيعية لسلسلة التنفس الميتوكوندرية 1).
يؤدي تطويل تكرار GAA إلى إسكات النسخ (transcriptional silencing)، مما يقلل من مستويات mRNA لـ FXN 1). يؤدي نقص الفرتاكسين إلى ما يلي:
تراكم الحديد في الميتوكوندريا: بسبب ضعف وظيفة معالجة الحديد.
انخفاض إنتاج ATP: بسبب خلل في وظيفة مركب سلسلة التنفس2).
يؤدي نقص الفرتاكسين إلى تغير مستويات الحديد داخل الخلايا، مما يجعل الخلايا العقدية الشبكية (RGC) عرضة للإجهاد التأكسدي. تظهر العلامات العينية لدى ما يصل إلى 30% من مرضى FA. يتأثر كل من جذع الدماغ والدوائر المخيخية والعصب البصري. يرتبط انخفاض سمك طبقة الألياف العصبية الشبكية (RNFL) ارتباطًا مباشرًا بانخفاض حدة البصر وحساسية التباين، ويرتبط العجز المبكر في المجال البصري الذي يتقدم بشكل متحد المركز من المحيط بفقدان RNFL المسجل بواسطة OCT.
يعد الخلل الوظيفي للميتوكوندريا السبب الأساسي لمرض السكري المرتبط بـ FRDA 2).
خلل وظيفي في خلايا بيتا: يؤدي ضعف إنتاج ATP إلى انخفاض إفراز الأنسولين، وفي النهاية إلى فقدان خلايا بيتا.
اضطراب تنظيم إفراز خلايا ألفا: فرط جلوكاجون الدم المتناقض أثناء ارتفاع السكر في الدم، وقصور استجابة الجلوكاجون أثناء انخفاض السكر في الدم 2).
آلية مقاومة الأنسولين: انخفاض امتصاص الجلوكوز بسبب ضعف الفسفرة التأكسدية في العضلات الهيكلية، وتراكم الدهون خارج الرحم في الكبد والعضلات بسبب اضطراب استقلاب الدهون، وضعف إشارات الأنسولين بسبب الالتهاب المزمن والإجهاد التأكسدي، وخلل في إنتاج الجلوكوز الكبدي بسبب الاعتلال العصبي الذاتي، وتدهور التمثيل الغذائي الثانوي المصاحب لعدم النشاط البدني وضمور العضلات بسبب الرنح 2).
يميل المرضى الذين لديهم حذف داخل الجين (Intragenic deletion) إلى ظهور مبكر للمرض، وتطور سريع، واعتلال عضلة القلب الشديد مقارنةً بمن لديهم توسع ثنائي الأليل (biallelic expansion)1). إذا أزال الحذف كودون البداية (start codon)، فإن إنتاج البروتين يتوقف تمامًا1).
Qلماذا تظهر أعراض عينية في مرض ترنح فريدريك؟
A
يؤدي نقص الفراتاكسين (frataxin) إلى خلل في استقلاب الحديد والإجهاد التأكسدي، مما يسبب تلف الخلايا العقدية الشبكية. يرتبط انخفاض سمك طبقة الألياف العصبية الشبكية (RNFL) ارتباطًا مباشرًا بانخفاض حدة البصر وحساسية التباين، ويؤدي إلى عيوب في المجال البصري تتقدم بشكل دائري من المحيط إلى المركز. كما يسبب تلف في جذع الدماغ والدوائر المخيخية اضطرابات في حركة العين مثل الرأرأة (nystagmus)، واضطراب حركات العين التتبعية، واضطراب قياس الحركات القافزة (saccades).
7. أحدث الأبحاث والتوجهات المستقبلية (تقارير في مرحلة البحث)
أبلغت التجربة السريرية التي أجراها Cooper et al. (2008) عن تحسن ملحوظ في درجة ICARS على مدى عامين باستخدام العلاج المشترك بـ CoQ10 وفيتامين E 2).
أظهرت دراسة تقاطعية عشوائية مضبوطة بالغفل أجراها Schöls et al. (2005) أن إعطاء L-كارنيتين يحسن بشكل ملحوظ إنتاج ATP الميتوكوندري 2).
أبلغ Sureshkumar et al. (2025) عن حالة لامرأة تبلغ من العمر 32 عامًا مصابة بداء السكري المرتبط بـ FA، تم إعطاؤها مزيجًا من العلاج بالأنسولين، سيتاجليبتين، L-كارنيتين، CoQ10، فيتامين E، فيتامينات عصبية، وإيميغليمين 2). تحسن HbA1c من 13.3% إلى 8.4% بعد 17 شهرًا، ثم إلى 6.9% بعد 19 شهرًا، وتحسنت درجة ICARS من 85 إلى 71 (بفارق 14 نقطة) (HOMA2-IR: 4.5→1.2، HOMA2-%B: 5→60، MAGE: 120→70 مغ/ديسيلتر، CV: 43%→34.9%). بينما يتوقع التاريخ الطبيعي وفقًا لـ SARA تدهورًا بحوالي 2.31 نقطة على مدى 3 سنوات، أظهرت الحالة انحرافًا إيجابيًا بحوالي 16 نقطة. تُعتبر هذه أول حالة يتم الإبلاغ عنها تحقق استقرارًا طويل الأمد لسكر الدم وتحسنًا في الرنح في وقت واحد.
يُعتقد أن الإيميغليمين يعمل من خلال تحسين نشاط سلسلة التنفس، وتقليل الإجهاد التأكسدي، وتعزيز تخليق ATP/NAD+ 2).
العلاج بالناقل الفيروسي المرتبط بالغدة (AAV) : تجربة سريرية لـ AAVrh.10hFXN تهدف إلى إدخال الجين الطبيعي FXN إلى القلب والجهاز العصبي، وقد أظهرت تحسنًا مبكرًا في تعبير FXN والعلامات المرضية (Munoz-Zuluaga et al. 2023)2).
CRISPR-Cas9: يهدف إلى إزالة تسلسلات تكرار GAA، ويجري دراسته في خلايا ونماذج فئران YG8R (Ouellet et al. 2017)2).
التحدي: من الضروري التحكم في الاستجابة المناعية وتطوير طريقة آمنة لتوصيل الجينات2).
أبلغ أغيليرا وآخرون (2023) عن تحديد حذف داخل جيني جديد يشمل المنطقة 5’UTR والإكسونات 1-2 من جين FXN عن طريق فحص عينات والدي مريض كان يُعتقد أنه يعاني من تمدد ثنائي الأليل 1). على الرغم من أنه تم الإبلاغ عن 10 حالات فقط من الحذف داخل الجيني في الأدبيات حتى الآن، إلا أنه يُشير إلى أن التكرار الفعلي قد يكون أعلى. يُعتبر توحيد التحليل الجيني التكميلي باستخدام MLPA وغيره وفحص عينات الوالدين من التحديات المستقبلية.
يشير التقرير عن أول حالة مؤكدة في غرب أفريقيا إلى الحاجة إلى دراسات أترابية واسعة النطاق عبر خلفيات عرقية وجغرافية متنوعة3). قد يصبح تحديد المتغيرات المعدلة للمرض أهدافًا علاجية مستقبلية.
Aguilera C, Esteve-Garcia A, Casasnovas C, et al. Novel intragenic deletion within the FXN gene in a patient with typical phenotype of Friedreich ataxia: may be more prevalent than we think? BMC Med Genomics. 2023;16:312.
Sureshkumar P, Kumar SS, Cheriyan J, Masood A. Friedreich Ataxia and Related Diabetes: Therapeutic Approach Targeting Mitochondrial Dysfunction. JCEM Case Rep. 2025;3:luaf215.
Cissé CAK, Cissé L, Ba HO, et al. Friedreich ataxia in a family from Mali, West Africa. Clin Case Rep. 2021;9:e04065.
انسخ نص المقال والصقه في مساعد الذكاء الاصطناعي الذي تفضله.
تم نسخ المقال إلى الحافظة
افتح أحد مساعدي الذكاء الاصطناعي أدناه والصق النص المنسوخ في مربع المحادثة.