全身神經學所見
運動失調性步態:全方位不穩定步態。伴隨測量障礙、肌張力低下、反覆拮抗運動不全、協同運動時間障礙1)。
深部肌腱反射消失:膝腱反射、跟腱反射消失是Harding診斷標準的必要項目。上肢反射消失也常見。部分病例出現反射亢進、痙攣、剪刀步態3)。
巴賓斯基徵:表現為伸性足底反應。
骨骼變形:脊柱側彎(早期出現)、凹足(pes cavus)、腰椎後彎3)。
全身併發症:肥厚型心肌病(最高63%)、糖尿病(5〜40%)2)。

弗里德賴希運動失調症(Friedreich Ataxia; FRDA)是一種影響中樞及周邊神經系統的進行性遺傳疾病。在白人族群中,它是最常見的體染色體隱性遺傳性運動失調症。
1863年,尼古拉斯·弗里德賴希報告了年輕發病的運動失調、脊柱側彎及家族性心臟變性。後來,皮埃爾·馬里將FRDA與其他運動失調症區分開來,確立了該疾病的概念。
流行病學上,地區差異很大,盛行率報告為1:20,000至1:750,000。歐洲人約為1:21,0001),全球估計為1:40,0002)至1:50,0003)。帶因者頻率約為1/701)。在法國南部、西班牙北部和愛爾蘭頻率較高,而在斯堪地納維亞和俄羅斯則較低。在撒哈拉以南非洲和東半球則更低,但在西非(馬利共和國的圖阿雷格族近親通婚家族)也有遺傳學確認的病例報告3)。
遺傳模式為體染色體隱性,男女發病率相同。典型發病年齡平均為15.5歲,多數在25歲前發病。8至15歲發病最常見2)。平均壽命為39歲,主要死因是心肌病2)。
在白人族群中最常見的遺傳性運動失調症,盛行率因地區而異,範圍為1:20,000至1:750,000。在歐洲約為每21,000人中有1人1),全球估計為每40,000至50,000人中有1人2)3)。帶因者頻率約為1/701)。
運動症狀的進展因人而異,但從初發症狀起平均約8年無法獨自行走,11~15年需要輪椅。有報告指出,GAA重複次數較長的患者在5年內就需要輪椅3)。
全身神經學所見
運動失調性步態:全方位不穩定步態。伴隨測量障礙、肌張力低下、反覆拮抗運動不全、協同運動時間障礙1)。
深部肌腱反射消失:膝腱反射、跟腱反射消失是Harding診斷標準的必要項目。上肢反射消失也常見。部分病例出現反射亢進、痙攣、剪刀步態3)。
巴賓斯基徵:表現為伸性足底反應。
骨骼變形:脊柱側彎(早期出現)、凹足(pes cavus)、腰椎後彎3)。
全身併發症:肥厚型心肌病(最高63%)、糖尿病(5〜40%)2)。
神經眼科所見
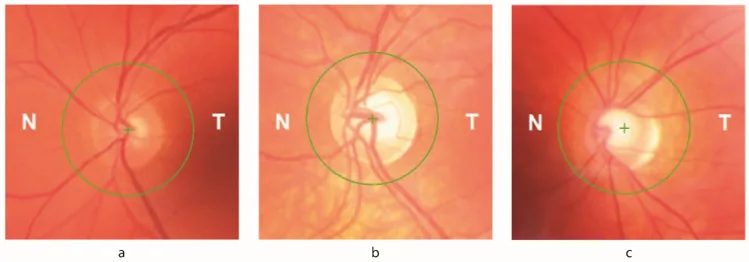
FRDA中最多30%的患者出現眼科徵象,但許多患者初期缺乏視覺自覺症狀。視神經萎縮、RNFL厚度減少、眼球運動障礙可通過客觀檢查檢測,並與視力及對比敏感度下降直接相關。建議定期進行眼科檢查。
FRDA的原因為第9號染色體上FXN(frataxin)基因內含子1中GAA三核苷酸重複序列的擴增。96%的病例為雙等位基因致病性擴增的純合子1)。其餘病例為GAA擴增與點突變,或GAA擴增與基因內/全基因缺失的複合雜合子1)。
GAA重複次數與疾病的關係如下所示。
| 分類 | GAA重複次數 |
|---|---|
| 正常 | 5~33次 |
| 中間(相當於帶因者) | 34~65次 |
| 致病性 | 66次以上(另報告為90次以上)1)3) |
GAA重複次數越長,發病年齡越早,且傾向於更嚴重。在一個等位基因重複次數極長(999/766)的病例中,患者在11歲發病,並在5年內需要輪椅(比一般的10年更快)3)。重複序列中的中斷(interruption)可能延遲發病年齡1)。
基因內缺失(intragenic deletion)合併GAA擴增的複合雜合子被認為比雙等位基因擴增更傾向於早期發病、快速進展和嚴重心肌病,但也有報告顯示典型病程的病例1)。
由於此疾病為體染色體隱性遺傳,近親結婚會增加發病風險2)3)。在歐洲裔人群中發生率較高。
由於PCR片段分析和TP-PCR無法檢測基因內缺失,因此看似雙等位擴增實際上可能是GAA擴增加上基因內缺失1)。透過MLPA(多重連接依賴性探針擴增)進行額外檢測以及親本樣本檢測,對於準確的遺傳諮詢至關重要。
臨床診斷採用Harding的標準。
| 類別 | 主要項目 |
|---|---|
| 必要項目 | 25歲前發病、進行性步態及四肢運動失調、膝腱及跟腱反射消失、軸突變性表現、構音障礙(發病5年後) |
| 附加發現(66%以上) | 脊柱側彎、下肢錐體路肌力減弱、上肢反射消失、粗大神經纖維感覺消失、心電圖異常 |
| 其他(低於50%) | 眼震、視神經萎縮、聽力喪失、遠端肌肉萎縮、弓形足、糖尿病 |
表現為感覺神經病變,特徵為感覺神經動作電位(SNAP)消失。軸索性感覺多發性神經病變的模式具有特徵性3)。
若以眼球運動障礙為主要症狀,需與以下疾病進行鑑別。
目前尚無根治性治療,治療核心為多專科協作的症狀治療與併發症管理。
神經內科與骨科
脊柱側彎管理:輕度至中度採用矯具治療;重度則考慮手術。
凹足管理:透過腓腸肌注射肉毒桿菌毒素、跟腱伸展來改善活動度。
復健:透過物理治療、職能治療、語言治療維持功能。
眼科
FRDA相關的糖尿病根源於粒線體功能障礙,因此藥物選擇需謹慎2)。
使用上述方案的病例報告顯示,糖化血紅蛋白從13.3%降至8.4%(17個月後),進一步降至6.9%(再19個月後),ICARS從85改善至71,改善了14分2)。
由於根本原因在於粒線體功能障礙,應避免使用metformin和thiazolidinediones2)。建議使用DPP-4抑制劑(如sitagliptin)和GLP-1類似物。L-肉鹼、輔酶Q10、維生素E等粒線體標靶輔助療法也可能有幫助2)。
Frataxin是一種位於粒線體內膜的蛋白質。它參與鐵代謝(鐵儲存、鐵硫簇組裝),對粒線體呼吸鏈的正常功能至關重要1)。
GAA重複序列的擴增會導致轉錄沉默(transcriptional silencing),降低FXN mRNA水平1)。Frataxin缺乏會導致以下情況。
由於弗拉塔辛缺乏導致細胞內鐵水平波動,視網膜神經節細胞(RGC)對氧化壓力變得脆弱。高達30%的FA患者出現眼科徵象。腦幹、小腦迴路及視神經均受損。RNFL厚度減少與視力及對比敏感度下降直接相關,從周邊向中心同心圓狀進展的早期視野缺損與OCT記錄的RNFL消失有關。
肥厚型心肌病導致心臟重量增加。心肌組織肉眼可見「大理石樣(marble-like)」外觀。
粒線體功能障礙是FRDA相關糖尿病的根本原因2)。
帶有基因內缺失的患者比雙等位基因擴增的患者有更早發病、快速進展及更嚴重心肌病的傾向1)。若缺失導致起始密碼子被移除,則蛋白質生成會完全消失1)。
Cooper等人(2008)的臨床試驗報告指出,CoQ10與維生素E的聯合療法在兩年期間顯著改善了ICARS評分2)。
Schöls等人(2005)的隨機安慰劑對照交叉試驗顯示,L-肉鹼給藥可顯著改善粒線體ATP生成2)。
Sureshkumar等人(2025)對一名患有FA相關糖尿病的32歲女性,給予胰島素療法、西格列汀、L-肉鹼、CoQ10、維生素E、神經營養維生素和imeglimin的組合治療2)。HbA1c從13.3%在17個月後改善至8.4%,再於19個月後改善至6.9%;ICARS從85降至71,改善14分(HOMA2-IR:4.5→1.2,HOMA2-%B:5→60,MAGE:120→70 mg/dL,CV:43%→34.9%)。根據SARA的自然病程,預測3年內惡化約2.31分,但該病例顯示出約16分的正向偏差。這是首次報告同時實現長期血糖穩定和運動失調改善的病例。
Imeglimin被認為通過改善呼吸鏈活性、減輕氧化壓力、促進ATP/NAD+合成而發揮作用2)。
Aguilera等人(2023)通過對疑似雙等位基因擴張患者的親本樣本檢測,鑑定出一個包含FXN基因5’UTR及外顯子1-2的新型基因內缺失1)。迄今文獻僅報告10例基因內缺失,但實際發生頻率可能更高。未來課題包括標準化使用MLPA等補充性基因分析及親本樣本檢測。
西非首次確診病例的報告顯示,需要在多樣化的民族和地理背景下進行大規模隊列研究3)。疾病修飾變異的鑑定可能成為未來治療的標靶。