Die Friedreich-Ataxie (FRDA) ist eine fortschreitende Erbkrankheit, die das zentrale und periphere Nervensystem betrifft. Sie ist die häufigste autosomal-rezessive Ataxie in der weißen Bevölkerung.
1863 berichtete Nikolaus Friedreich über juvenile Ataxie, Skoliose und familiäre Herzdegeneration. Später unterschied Pierre Marie die Friedreich-Ataxie von anderen Ataxien und etablierte das Krankheitskonzept.
Epidemiologisch gibt es große regionale Unterschiede, die Prävalenz wird mit 1:20.000 bis 1:750.000 angegeben. Bei Europäern liegt sie bei etwa 1:21.0001), weltweit wird sie auf 1:40.0002) bis 1:50.0003) geschätzt. Die Trägerfrequenz wird mit etwa 1/70 angegeben1). Die Häufigkeit ist in Südfrankreich, Nordspanien und Irland hoch, in Skandinavien und Russland niedrig. In Subsahara-Afrika und der östlichen Hemisphäre ist sie noch geringer, aber es wurden auch genetisch bestätigte Fälle in Westafrika (bei konsanguinen Tuareg-Familien in Mali) berichtet3).
Der Erbgang ist autosomal-rezessiv, und die Inzidenzrate ist bei Männern und Frauen gleich. Das typische Erkrankungsalter liegt im Durchschnitt bei 15,5 Jahren, die meisten Fälle treten vor dem 25. Lebensjahr auf. Ein Beginn zwischen 8 und 15 Jahren ist häufig2). Die durchschnittliche Lebenserwartung beträgt 39 Jahre, und die Haupttodesursache ist die Kardiomyopathie2).
QWie häufig tritt die Friedreich-Ataxie auf?
A
Es ist die häufigste hereditäre Ataxie in der weißen Bevölkerung, mit einer Prävalenz von 1:20.000 bis 1:750.000 je nach Region. In Europa betrifft sie etwa 1 von 21.000 Menschen1), weltweit wird sie auf 1 von 40.000 bis 50.000 geschätzt2)3). Die Trägerfrequenz wird mit etwa 1/70 angegeben1).
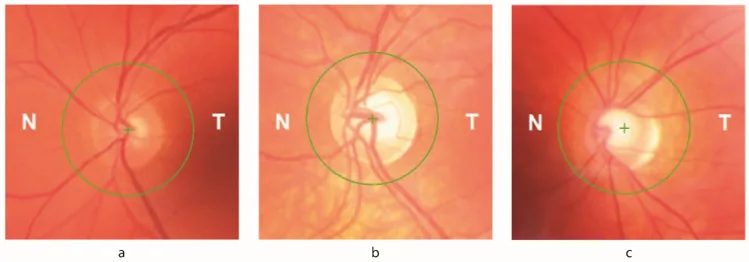
Petya Bogdanova-Mihaylova, Helena Maria Plapp, Hongying Chen et al. Longitudinal Assessment Using Optical Coherence Tomography in Patients with Friedreich’s Ataxia. Tomography. 2021 Dec 8; 7(4):915. Figure 1. PMCID: PMC8706975. License: CC BY.
Gangstörung : häufigstes Erstsymptom. Beginnt mit unsicherem Gang in alle Richtungen.
Ungeschicklichkeit und Tremor der oberen Extremitäten : in einigen Fällen beginnt es mit Handtremor3).
Dysarthrie : tritt oft innerhalb von 10–15 Jahren nach Krankheitsbeginn auf.
Sensorische Symptome : Verlust der Tiefensensibilität (Propriozeption).
Visuelle Symptome: Bei einigen Patienten kommt es zu einer verminderten Sehschärfe und Kontrastempfindlichkeit, aber die Mehrheit weist im Frühstadium keine subjektiven visuellen Symptome auf.
Das Fortschreiten der motorischen Symptome variiert von Person zu Person, aber im Durchschnitt wird etwa 8 Jahre nach dem Auftreten der ersten Symptome das selbstständige Gehen unmöglich, und nach 11–15 Jahren ist ein Rollstuhl erforderlich. Bei einer hohen Anzahl von GAA-Wiederholungen wurde berichtet, dass bereits nach 5 Jahren ein Rollstuhl benötigt wurde 3).
Klinische Befunde (vom Arzt bei der Untersuchung festgestellte Befunde)
Ataktischer Gang: in alle Richtungen unsicherer Gang. Begleitet von Dysmetrie, Hypotonie, Dysdiadochokinese und Störung der Bewegungskoordination1).
Fehlen der tiefen Sehnenreflexe : Das Fehlen des Patellar- und Achillessehnenreflexes ist ein obligates Kriterium der Harding-Diagnosekriterien. Auch das Fehlen der Reflexe der oberen Extremitäten tritt häufig auf. In einigen Fällen können Hyperreflexie, Spastik oder Scherengang auftreten3).
Babinski-Zeichen : Tritt als extensorische Plantarantwort auf.
Nystagmus : Aufgrund einer Schädigung der Kleinhirn-Hirnstamm-Schaltkreise. Einschließlich blickrichtungsabhängigem Nystagmus.
Square-wave jerks : Unwillkürliche, impulsive Augenbewegungen, die während der Fixation auftreten.
Störung der glatten Verfolgung : Die Zielverfolgung wird treppenförmig (Pursuit-Anomalie).
Sakkadische Dysmetrie : Es treten überschießende oder unterschreitende Sakkaden auf.
Optikusatrophie : Im Fundusbefund nachweisbar. Bei bis zu 30 % der Patienten treten ophthalmologische Zeichen auf.
RNFL-Dickenabnahme : Mittels OCT nachweisbar. Direkt korreliert mit verminderter Sehschärfe und Kontrastempfindlichkeit.
Gesichtsfeldausfall : Initialer Gesichtsfeldausfall, der konzentrisch von der Peripherie fortschreitet. Verbunden mit dem im OCT dokumentierten RNFL-Verlust.
QTreten die Augensymptome immer auf?
A
Bei FRDA treten bei bis zu 30 % der Patienten ophthalmologische Anzeichen auf, aber die meisten Patienten haben anfangs keine subjektiven Sehstörungen. Optikusatrophie, verminderte RNFL-Dicke und Augenbewegungsstörungen werden durch objektive Tests nachgewiesen und korrelieren direkt mit verminderter Sehschärfe und Kontrastempfindlichkeit. Regelmäßige augenärztliche Untersuchungen werden empfohlen.
Die Ursache der FRDA ist eine Verlängerung der GAA-Triplett-Wiederholung im Intron 1 des FXN-Gens (Frataxin) auf Chromosom 9. 96% der Fälle sind homozygot für eine biallelische pathogene Expansion1). Die restlichen Fälle sind zusammengesetzte Heterozygote mit einer GAA-Expansion und einer Punktmutation oder einer GAA-Expansion und einer intragene/ganzgen-Deletion1).
Der Zusammenhang zwischen der Anzahl der GAA-Wiederholungen und der Erkrankung ist unten dargestellt.
Klassifikation
GAA-Wiederholungsanzahl
Normal
5–33
Intermediär (Trägerstatus)
34–65
Pathologisch
66 oder mehr Wiederholungen (nach anderen Berichten 90 oder mehr) 1)3)
Je länger die GAA-Wiederholungsanzahl, desto früher das Erkrankungsalter und desto schwerer der Verlauf. Bei einem Fall mit extrem langer Allel-Wiederholungszahl von 999/766 begann die Erkrankung im Alter von 11 Jahren und innerhalb von 5 Jahren war ein Rollstuhl erforderlich (schneller als die üblichen 10 Jahre) 3). Unterbrechungen innerhalb der Wiederholungssequenz können das Erkrankungsalter verzögern 1).
Komposite Heterozygote mit intragener Deletion + GAA-Expansion neigen zu früherem Beginn, schnellerem Fortschreiten und schwererer Kardiomyopathie als biallelische Expansionen, es wurden jedoch auch Fälle mit typischem Verlauf berichtet 1).
Blutsverwandtschaft erhöht aufgrund des autosomal-rezessiven Erbgangs das Erkrankungsrisiko2)3). Die Erkrankung tritt häufiger bei Bevölkerungsgruppen europäischer Abstammung auf.
QWenn der Gentest einen homozygoten Status ergibt, besteht die Möglichkeit einer compound-heterozygoten Konstellation?
A
Die PCR-Fragmentanalyse und die TP-PCR können keine intragenischen Deletionen nachweisen. Daher kann eine scheinbare biallelische Expansion tatsächlich eine GAA-Expansion in Kombination mit einer intragenischen Deletion sein1). Zusätzliche Tests mittels MLPA (Multiplex Ligation-dependent Probe Amplification) und die Untersuchung von Elternproben sind für eine präzise genetische Beratung unerlässlich.
Für die klinische Diagnose werden die Harding-Kriterien verwendet.
Kategorie
Hauptpunkte
Erforderliche Kriterien
Beginn vor dem 25. Lebensjahr, progressive Gang- und Extremitätenataxie, Fehlen von Patellar- und Achillessehnenreflexen, axonale Degeneration, Dysarthrie (ab 5 Jahren nach Beginn)
Zusätzliche Befunde (≥66%)
Skoliose, pyramidale Schwäche der unteren Extremitäten, Areflexie der oberen Extremitäten, sensorischer Verlust dicker Nervenfasern, EKG-Anomalien
PCR + TP-PCR : Amplifikation der GAA-Wiederholung in Intron 1 zum Nachweis des expandierten Allels.
MLPA (Multiplex Ligation-dependent Probe Amplification) : notwendig für die Diagnose von Deletionen und Duplikationen, die durch Fragmentanalyse oder TP-PCR nicht nachweisbar sind1). Wird hinzugefügt, um die Möglichkeit auszuschließen, dass eine scheinbare biallelische Expansion ein zusammengesetzter Heterozygot ist.
Southern-Blot-Analyse : in Kombination mit PCR ist eine Diagnose mit einer Genauigkeit von über 99 % möglich3).
Untersuchung der Elternproben : unerlässlich für eine genaue genetische Beratung1).
Zeigt eine sensorische Neuropathie, die durch das Verschwinden des sensorischen Nervenaktionspotentials (SNAP) gekennzeichnet ist. Das Muster einer axonalen sensorischen Polyneuropathie ist charakteristisch3).
Elektrookulographie (EOG) : Wird zur qualitativen und quantitativen Analyse von Augenbewegungen verwendet. Bewertet visuell geführte Sakkaden (Latenz und Amplitude), Verfolgung (Verstärkung bei verschiedenen Zielgeschwindigkeiten), VOR (Verstärkung bei verschiedenen Kopfwinkelgeschwindigkeiten) und Fixation (Nystagmus-Wellenformanalyse, Rechteckwellenlatenz).
Optische Kohärenztomographie (OCT) : Quantitative Messung der Dicke der retinalen Nervenfaserschicht (RNFL). Korrelation mit verminderter Sehschärfe und Kontrastempfindlichkeit kann bestätigt werden.
Gesichtsfelduntersuchung: Beurteilung des von der Peripherie konzentrisch fortschreitenden Defektmusters.
Fundusuntersuchung: Prüfung auf Vorliegen einer Optikusatrophie.
EKG und Echokardiographie: Beurteilung von Kardiomyopathie, linksventrikulärer Wanddicke, Ejektionsfraktion und diastolischer Funktion2). Regelmäßige Überwachung erforderlich.
Blutzuckerbezogene Tests2): Nüchternblutzucker, HbA1c, C-Peptid, Nüchterninsulin, Pankreasinsel-Autoantikörper (GAD65, IA-2, ZnT8), HOMA2-IR, HOMA2-%B zur Früherkennung und Klassifikation des Diabetes.
Klinische Bewertungsskalen: FARS (Friedreich Ataxia Rating Scale), ICARS, SARA zur quantitativen Bewertung neurologischer Symptome2). Für SARA wurde eine jährliche Verschlechterung von 0,77 Punkten (SE 0,06) im natürlichen Verlauf berichtet2).
MRT von Gehirn und Rückenmark: Beurteilung der Kleinhirnatrophie (in einigen Fällen 3 Jahre nach Beginn aufgetreten3)) und degenerativer Veränderungen des Rückenmarks.
Derzeit gibt es keine kurative Therapie; die Behandlung konzentriert sich auf symptomatische Therapie und Komplikationsmanagement durch multidisziplinäre Zusammenarbeit.
Skoliose-Management : Bei leichter bis mittelschwerer Skoliose Korsetttherapie; bei schwerer Skoliose Operation in Betracht ziehen.
Hohlfuß-Management : Botulinumtoxin-Injektion in den Gastrocnemius-Muskel und Dehnung der Achillessehne zur Verbesserung der Beweglichkeit.
Rehabilitation : Erhalt der Funktionen durch Physiotherapie, Ergotherapie und Logopädie.
Augenheilkunde
Sehbehindertenversorgung : Für Patienten mit Sehstörungen aufgrund von Optikusatrophie oder Netzhautatrophie Bereitstellung von Lupen, Lichtanpassung und Lebensberatung.
Symptomatische Behandlung von Augenbewegungsstörungen: Bei lageabhängigem Nystagmus werden Prismenbrillen verwendet (beidseits gleiche Prismenstärke in die Richtung, die die Position verschlechtert). Bei vertikalem Nystagmus, periodisch wechselndem Nystagmus und sakkadischen Augenbewegungen wird ein GABA_B-Agonist verabreicht.
Verschreibungsbeispiel (GABA_B-Agonist): 3–6 Tabletten Gabaron (5 mg), 1–3 mal täglich.
Da dem mit FRDA assoziierten Diabetes eine mitochondriale Dysfunktion zugrunde liegt, ist bei der Medikamentenauswahl Vorsicht geboten2).
Zu vermeidende Medikamente: Metformin und Thiazolidindione hemmen den mitochondrialen Komplex I, daher vermeiden2). Sulfonylharnstoffe bergen ein Risiko für Betazellstress und Hypoglykämie.
Empfohlene blutzuckersenkende Medikamente: DPP-4-Hemmer (z. B. Sitagliptin 100 mg/Tag) und GLP-1-Analoga werden bevorzugt2). Imeglimin (500 mg x 2/Tag) könnte als neues blutzuckersenkendes Medikament, das auf die Mitochondrienfunktion abzielt, nützlich sein2).
Insulintherapie : Beginn mit etwa 0,5 U/kg/Tag2).
Mitochondrien-gerichtete adjuvante Therapie: Die Kombination von L-Carnitin 500 mg/Tag, CoQ10 100 mg/Tag und Vitamin E 400 IE/Tag wurde berichtet2).
Im Fallbericht mit dem obigen Regime verbesserte sich der HbA1c von 13,3 % auf 8,4 % (nach 17 Monaten) und weiter auf 6,9 % (nach weiteren 19 Monaten), und der ICARS sank von 85 auf 71, eine Verbesserung um 14 Punkte2).
QWelche Behandlung ist für Diabetes im Zusammenhang mit Friedreich-Ataxie geeignet?
A
Aufgrund der zugrunde liegenden mitochondrialen Dysfunktion sollten Metformin und Thiazolidindione vermieden werden2). DPP-4-Hemmer (wie Sitagliptin) und GLP-1-Analoga werden empfohlen. Mitochondrien-gerichtete adjuvante Therapien wie L-Carnitin, CoQ10 und Vitamin E können ebenfalls nützlich sein2).
6. Pathophysiologie und detaillierter Krankheitsmechanismus
Frataxin ist ein Protein, das in der inneren Mitochondrienmembran lokalisiert ist. Es ist am Eisenstoffwechsel (Eisenspeicherung und Zusammenbau von Eisen-Schwefel-Clustern) beteiligt und für die normale Funktion der mitochondrialen Atmungskette unerlässlich1).
Die Verlängerung der GAA-Wiederholungssequenz führt zu transkriptionellem Silencing (transcriptional silencing) und verringert die FXN-mRNA-Spiegel1). Ein Frataxin-Mangel führt zu Folgendem.
Mitochondriale Eisenakkumulation: aufgrund einer beeinträchtigten Eisenverarbeitung.
Verringerte ATP-Produktion: Funktionsstörung der Atmungskettenkomplexe2).
Spinalganglion : klein und atrophisch, die hintere Wurzel ist dünn und grau gefärbt.
Rückenmark: Durchmesserverringerung über die gesamte Länge, besonders ausgeprägt im thorakalen Bereich. Progressive Degeneration der kortikospinalen Bahnen, der spinocerebellären Bahnen und der Hinterstränge.
Frataxin-Mangel führt zu Schwankungen des intrazellulären Eisenspiegels, wodurch retinale Ganglienzellen (RGC) anfälliger für oxidativen Stress werden. Bis zu 30 % der FA-Patienten zeigen ophthalmologische Anzeichen. Sowohl die Hirnstamm-Kleinhirn-Schaltkreise als auch der Sehnerv sind betroffen. Die Abnahme der RNFL-Dicke korreliert direkt mit einer Verschlechterung der Sehschärfe und des Kontrastsehens, und frühe Gesichtsfeldausfälle, die konzentrisch von der Peripherie fortschreiten, sind mit einem im OCT dokumentierten RNFL-Verlust verbunden.
Die hypertrophe Kardiomyopathie führt zu einer Zunahme des Herzgewichts. Das Myokardgewebe kann makroskopisch ein „marmorartiges“ (marble-like) Aussehen aufweisen.
Mitochondriale Dysfunktion ist die zugrunde liegende Ursache des FRDA-assoziierten Diabetes2).
β-Zell-Dysfunktion : Verminderte ATP-Produktion führt zu reduzierter Insulinsekretion und schließlich zum Verlust der β-Zellen.
Dysregulation der α-Zell-Sekretion : Paradoxe Hyperglukagonämie bei Hyperglykämie und unzureichende Glukagonantwort bei Hypoglykämie2).
Mechanismen der Insulinresistenz : Verminderte Glukoseaufnahme aufgrund gestörter oxidativer Phosphorylierung im Skelettmuskel, ektopische Fettablagerung in Leber und Muskel aufgrund von Lipidstoffwechselstörungen, beeinträchtigte Insulinsignalisierung durch chronische Entzündung und oxidativen Stress, abnorme hepatische Glukoseproduktion aufgrund autonomer Neuropathie, und sekundäre metabolische Verschlechterung durch körperliche Inaktivität und Muskelatrophie infolge der Ataxie2).
Träger einer intragene Deletion neigen zu früherem Beginn, schnellerem Fortschreiten und schwererer Kardiomyopathie im Vergleich zur biallelischen Expansion1). Wenn die Deletion das Startcodon entfernt, wird die Proteinproduktion vollständig aufgehoben1).
QWarum verursacht die Friedreich-Ataxie Augensymptome?
A
Der durch Frataxin-Mangel verursachte abnorme Eisenstoffwechsel und oxidativer Stress schädigen die retinalen Ganglienzellen. Die Abnahme der RNFL-Dicke korreliert direkt mit einer verminderten Sehschärfe und Kontrastempfindlichkeit, was zu einem von der Peripherie konzentrisch fortschreitenden Gesichtsfeldausfall führt. Darüber hinaus verursachen Schäden an den Hirnstamm- und Kleinhirnschaltkreisen Augenbewegungsstörungen wie Nystagmus, Störungen der Blickfolgebewegungen und Sakkadenmessungsstörungen.
7. Aktuelle Forschung und zukünftige Perspektiven (Berichte aus der Forschungsphase)
Die klinische Studie von Cooper et al. (2008) berichtete über eine signifikante Verbesserung des ICARS-Scores über zwei Jahre durch eine Kombinationstherapie mit CoQ10 und Vitamin E2).
Die randomisierte, placebokontrollierte Crossover-Studie von Schöls et al. (2005) zeigte, dass die Gabe von L-Carnitin die mitochondriale ATP-Produktion signifikant verbesserte2).
Sureshkumar et al. (2025) behandelten eine 32-jährige Frau mit FA-assoziiertem Diabetes mit einer Kombination aus Insulintherapie, Sitagliptin, L-Carnitin, CoQ10, Vitamin E, neurotropen Vitaminen und Imeglimin2). Der HbA1c-Wert verbesserte sich von 13,3 % auf 8,4 % nach 17 Monaten und weiter auf 6,9 % nach 19 Monaten, und der ICARS-Score sank von 85 auf 71, eine Verbesserung um 14 Punkte (HOMA2-IR: 4,5→1,2; HOMA2-%B: 5→60; MAGE: 120→70 mg/dL; CV: 43 %→34,9 %). Während der natürliche Verlauf nach SARA eine Verschlechterung um etwa 2,31 Punkte über drei Jahre vorhersagt, zeigte sich eine positive Abweichung von etwa 16 Punkten. Dies ist der erste berichtete Fall einer gleichzeitigen langfristigen Blutzuckerstabilisierung und Verbesserung der Ataxie.
Es wird angenommen, dass Imeglimin durch Verbesserung der Atmungskettenaktivität, Reduzierung von oxidativem Stress und Förderung der ATP/NAD+-Synthese wirkt2).
AAV-Vektor-Therapie : Die klinische Studie AAVrh.10hFXN zur Einführung des normalen FXN-Gens in Herz und Nervensystem ist im Gange und hat eine frühe Verbesserung der FXN-Expression und Krankheitsmarker gezeigt (Munoz-Zuluaga et al. 2023)2).
CRISPR-Cas9 : Zur Entfernung von GAA-Wiederholungssequenzen wird an YG8R-abgeleiteten Zellen und Mausmodellen geforscht (Ouellet et al. 2017)2).
Herausforderung : Die Kontrolle der Immunantwort und die Etablierung einer sicheren Gentransfermethode sind erforderlich2).
Verbesserung der Genauigkeit der genetischen Diagnostik
Aguilera et al. (2023) identifizierten eine neue intragene Deletion, die die 5’UTR und die Exons 1-2 des FXN-Gens umfasst, durch Untersuchung von Elternproben eines Patienten, bei dem eine biallelische Expansion vermutet wurde 1). Bisher wurden in der Literatur nur 10 intragene Deletionen berichtet, aber es wird vermutet, dass sie tatsächlich häufiger vorkommen. Die Standardisierung ergänzender genetischer Analysen mittels MLPA und von Elternprobenuntersuchungen wird als zukünftige Herausforderung angesehen.
Der Bericht des ersten bestätigten Falls in Westafrika zeigt die Notwendigkeit groß angelegter Kohortenstudien mit unterschiedlichen ethnischen und geografischen Hintergründen3). Die Identifizierung krankheitsmodifizierender Varianten könnte zukünftige therapeutische Ziele darstellen.
Aguilera C, Esteve-Garcia A, Casasnovas C, et al. Novel intragenic deletion within the FXN gene in a patient with typical phenotype of Friedreich ataxia: may be more prevalent than we think? BMC Med Genomics. 2023;16:312.
Sureshkumar P, Kumar SS, Cheriyan J, Masood A. Friedreich Ataxia and Related Diabetes: Therapeutic Approach Targeting Mitochondrial Dysfunction. JCEM Case Rep. 2025;3:luaf215.
Cissé CAK, Cissé L, Ba HO, et al. Friedreich ataxia in a family from Mali, West Africa. Clin Case Rep. 2021;9:e04065.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.