L’atassia di Friedreich (FRDA) è una malattia genetica progressiva che colpisce il sistema nervoso centrale e periferico. È l’atassia autosomica recessiva più comune nella popolazione bianca.
Nel 1863, Nikolaus Friedreich riportò atassia giovanile, scoliosi e degenerazione cardiaca familiare. Successivamente, Pierre Marie distinse l’atassia di Friedreich dalle altre atassie, stabilendo il concetto di malattia.
Dal punto di vista epidemiologico, ci sono grandi differenze regionali, con una prevalenza riportata da 1:20.000 a 1:750.000. Negli europei è di circa 1:21.0001), mentre a livello mondiale si stima da 1:40.0002) a 1:50.0003). La frequenza dei portatori è di circa 1/701). La frequenza è alta nel sud della Francia, nel nord della Spagna e in Irlanda, mentre è bassa in Scandinavia e Russia. È ancora più bassa nell’Africa subsahariana e nell’emisfero orientale, ma sono stati riportati casi geneticamente confermati anche in Africa occidentale (in famiglie tuareg consanguinee in Mali)3).
La modalità di trasmissione è autosomica recessiva e il tasso di incidenza è uguale tra maschi e femmine. L’età tipica di esordio è in media di 15,5 anni, e la maggior parte dei casi esordisce prima dei 25 anni. L’esordio è frequente tra gli 8 e i 15 anni2). L’aspettativa di vita media è di 39 anni e la causa principale di morte è la cardiomiopatia2).
QCon quale frequenza si verifica l'atassia di Friedreich?
A
È l’atassia ereditaria più frequente nella popolazione bianca, con una prevalenza che varia da 1:20.000 a 1:750.000 a seconda della regione. In Europa colpisce circa 1 persona su 21.0001), mentre a livello mondiale si stima che colpisca 1 persona su 40.000-50.0002)3). La frequenza dei portatori è di circa 1/701).
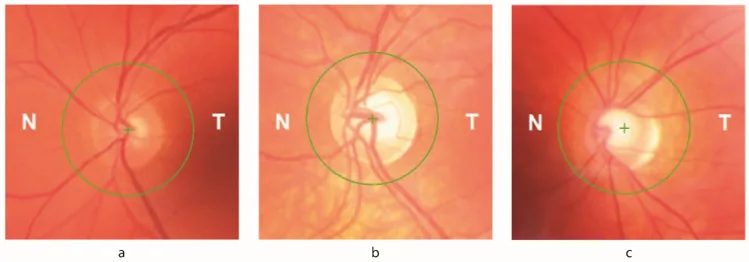
Petya Bogdanova-Mihaylova, Helena Maria Plapp, Hongying Chen et al. Longitudinal Assessment Using Optical Coherence Tomography in Patients with Friedreich’s Ataxia. Tomography. 2021 Dec 8; 7(4):915. Figure 1. PMCID: PMC8706975. License: CC BY.
Disturbo dell’andatura : sintomo iniziale più comune. Inizia con un’andatura instabile in tutte le direzioni.
Impaccio e tremore degli arti superiori : in alcuni casi esordisce con tremore alle mani3).
Disartria : spesso compare entro 10-15 anni dall’esordio della malattia.
Sintomi sensoriali : perdita della sensibilità profonda (propriocezione).
Sintomi visivi: alcuni pazienti presentano una riduzione dell’acuità visiva e della sensibilità al contrasto, ma la maggioranza non presenta sintomi visivi soggettivi nella fase iniziale.
La progressione dei sintomi motori varia da persona a persona, ma in media, circa 8 anni dopo la comparsa dei primi sintomi, la deambulazione autonoma diventa impossibile e dopo 11-15 anni è necessaria una sedia a rotelle. Nei casi con un numero elevato di ripetizioni GAA, è stato riportato che la sedia a rotelle era necessaria dopo 5 anni 3).
Reperti clinici (segni riscontrati dal medico durante la visita)
Andatura atassica: instabilità della marcia in tutte le direzioni. Associata a dismetria, ipotonia, disdiadococinesia e alterazione della coordinazione motoria1).
Abolizione dei riflessi osteotendinei profondi : L’abolizione del riflesso rotuleo e achilleo è un criterio obbligatorio nei criteri diagnostici di Harding. L’abolizione dei riflessi degli arti superiori è anch’essa frequente. Alcuni casi presentano iperreflessia, spasticità o andatura a forbice3).
Segno di Babinski : Si manifesta come risposta plantare in estensione.
Nistagmo : Dovuto a danno dei circuiti cerebellari e del tronco encefalico. Include nistagmo dipendente dalla posizione dello sguardo.
Square-wave jerks : Movimenti oculari involontari impulsivi che si verificano durante la fissazione.
Disturbo dell’inseguimento lento : L’inseguimento del bersaglio diventa a scatti (anomalia di inseguimento).
Dismetria saccadica (saccadic dysmetria) : Comparsa di saccadi eccessive o insufficienti.
Atrofia ottica : Evidenziata all’esame del fondo oculare. Segni oftalmologici compaiono fino al 30% dei pazienti.
Riduzione dello spessore dello strato delle fibre nervose retiniche (RNFL) : Rilevata mediante OCT. Correlata direttamente alla riduzione dell’acuità visiva e della sensibilità al contrasto.
Difetto del campo visivo : Difetto del campo visivo iniziale che progredisce concentricamente dalla periferia. Associato alla perdita di RNFL documentata all’OCT.
QI sintomi oculari compaiono sempre?
A
Nella FRDA, fino al 30% dei pazienti presenta segni oftalmologici, ma la maggior parte dei pazienti inizialmente non ha sintomi visivi soggettivi. L’atrofia ottica, la riduzione dello spessore dello RNFL e i disturbi dei movimenti oculari vengono rilevati con esami obiettivi e sono direttamente correlati a una riduzione dell’acuità visiva e della sensibilità al contrasto. Si raccomanda una visita oculistica regolare.
La causa della FRDA è l’espansione della tripletta GAA nell’introne 1 del gene FXN (frataxina) sul cromosoma 9. Il 96% dei casi è omozigote per un’espansione patogena biallelica1). I restanti casi sono eterozigoti composti con un’espansione GAA e una mutazione puntiforme, o un’espansione GAA e una delezione intragenica/dell’intero gene1).
La relazione tra il numero di ripetizioni GAA e la malattia è mostrata di seguito.
Classificazione
Numero di ripetizioni GAA
Normale
5–33
Intermedio (portatore)
34–65
Patologico
66 o più ripetizioni (90 o più secondo altri rapporti) 1)3)
Maggiore è il numero di ripetizioni GAA, più precoce è l’età di insorgenza e più grave è la malattia. In un caso con un numero di ripetizioni alleliche estremamente lungo di 999/766, la malattia è iniziata a 11 anni e la sedia a rotelle è stata necessaria entro 5 anni (più rapido dei soliti 10 anni) 3). Le interruzioni all’interno della sequenza ripetuta possono ritardare l’età di insorgenza 1).
Gli eterozigoti composti con delezione intragenica + espansione GAA tendono a un esordio più precoce, progressione più rapida e cardiomiopatia più grave rispetto all’espansione biallelica, ma sono stati riportati anche casi con decorso tipico 1).
I matrimoni tra consanguinei aumentano il rischio di sviluppare la malattia a causa della sua natura autosomica recessiva2)3). La patologia è più frequente nelle popolazioni di origine europea.
QSe il test genetico mostra uno stato omozigote, è possibile che si tratti in realtà di un eterozigote composto?
A
L’analisi dei frammenti PCR e la TP-PCR non rilevano delezioni intrageniche, pertanto un’apparente espansione biallelica potrebbe in realtà essere una combinazione di espansione GAA e delezione intragenica1). Test aggiuntivi mediante MLPA (Multiplex Ligation-dependent Probe Amplification) e l’analisi dei campioni parentali sono essenziali per una consulenza genetica accurata.
Per la diagnosi clinica vengono utilizzati i criteri di Harding.
Categoria
Elementi principali
Criteri obbligatori
Esordio prima dei 25 anni, atassia progressiva del cammino e degli arti, assenza dei riflessi patellari e achillei, degenerazione assonale, disartria (dopo 5 anni dall’esordio)
Reperti aggiuntivi (≥66%)
Scoliosi, debolezza piramidale degli arti inferiori, areflessia degli arti superiori, perdita sensoriale delle fibre nervose spesse, anomalie ECG
PCR + TP-PCR : Amplificazione della ripetizione GAA nell’introne 1 per rilevare l’allele espanso.
MLPA (Multiplex Ligation-dependent Probe Amplification) : necessaria per diagnosticare delezioni e duplicazioni non rilevabili mediante analisi dei frammenti o TP-PCR1). Aggiunto per escludere la possibilità che un’apparente espansione biallelica sia un eterozigote composto.
Analisi Southern blot : in combinazione con la PCR consente una diagnosi con una precisione superiore al 99%3).
Esame dei campioni parentali : indispensabile per una consulenza genetica accurata1).
Mostra una neuropatia sensoriale caratterizzata dalla scomparsa del potenziale d’azione del nervo sensitivo (SNAP). Il pattern di una polineuropatia sensoriale assonale è caratteristico3).
Elettro-oculografia (EOG) : utilizzata per l’analisi qualitativa e quantitativa dei movimenti oculari. Valuta le saccadi visivamente guidate (latenza e ampiezza), l’inseguimento (guadagno per diverse velocità del bersaglio), il VOR (guadagno per diverse velocità angolari della testa) e la fissazione (analisi della forma d’onda del nistagmo, latenza delle onde quadre).
Tomografia a coerenza ottica (OCT) : misurazione quantitativa dello spessore dello strato di fibre nervose retiniche (RNFL). Consente di confermare la correlazione con la riduzione dell’acuità visiva e della sensibilità al contrasto.
Esame del campo visivo: valutare il pattern di difetto che progredisce concentricamente dalla periferia.
Esame del fondo oculare: verificare la presenza di atrofia del nervo ottico.
ECG ed ecocardiografia: valutano cardiomiopatia, spessore della parete ventricolare sinistra, frazione di eiezione e funzione diastolica2). È necessario un monitoraggio regolare.
Test correlati alla glicemia2): glicemia a digiuno, HbA1c, peptide C, insulina a digiuno, autoanticorpi delle isole pancreatiche (GAD65, IA-2, ZnT8), HOMA2-IR, HOMA2-%B per la diagnosi precoce e la classificazione del diabete.
Scale di valutazione clinica: FARS (Friedreich Ataxia Rating Scale), ICARS, SARA per la valutazione quantitativa dei sintomi neurologici2). Con SARA è stato riportato un peggioramento annuale di 0,77 punti (SE 0,06) nel decorso naturale2).
Risonanza magnetica cerebrale e spinale: valutazione dell’atrofia cerebellare (comparsa in alcuni casi 3 anni dopo l’esordio3)) e dei segni degenerativi del midollo spinale.
Attualmente non esiste una terapia curativa; il trattamento si basa sulla terapia sintomatica e sulla gestione delle complicanze attraverso un approccio multidisciplinare.
Gestione della scoliosi : per i casi lievi o moderati, terapia con busto ortopedico; per i casi gravi, considerare l’intervento chirurgico.
Gestione del piede cavo : iniezione di tossina botulinica nel muscolo gastrocnemio e stretching del tendine d’Achille per migliorare la mobilità.
Riabilitazione : mantenimento delle funzioni attraverso fisioterapia, terapia occupazionale e logopedia.
Oftalmologia
Cura per ipovisione : per i pazienti con sintomi visivi dovuti ad atrofia del nervo ottico o retinica, fornire lenti d’ingrandimento, regolazione dell’illuminazione e consigli per la vita quotidiana.
Trattamento sintomatico dei disturbi oculomotori: Per il nistagmo dipendente dalla posizione degli occhi si utilizzano occhiali prismatici (aggiungendo la stessa potenza prismatica a entrambi gli occhi nella direzione che peggiora la posizione). Per il nistagmo verticale, il nistagmo alternante periodico e i movimenti oculari saccadici si somministra un agonista GABA_B.
Esempio di prescrizione (agonista GABA_B): 3-6 compresse di Gabaron (5 mg), 1-3 volte al giorno.
Il diabete associato alla FRDA è dovuto a una disfunzione mitocondriale, quindi la scelta dei farmaci richiede attenzione2).
Farmaci da evitare: Metformina e tiazolidinedioni inibiscono il complesso I mitocondriale, quindi evitarli2). Le sulfoniluree comportano rischio di stress delle cellule beta e ipoglicemia.
Farmaci ipoglicemizzanti raccomandati: Gli inibitori della DPP-4 (es. sitagliptin 100 mg/die) e gli analoghi del GLP-1 sono preferiti2). L’imeglimina (500 mg x 2/die) potrebbe essere utile come nuovo farmaco ipoglicemizzante che mira alla funzione mitocondriale2).
Terapia insulinica : iniziare con circa 0,5 U/kg/die2).
Terapia adiuvante mirata ai mitocondri: è stata riportata la combinazione di L-carnitina 500 mg/die, CoQ10 100 mg/die e vitamina E 400 UI/die2).
Nel rapporto del caso che utilizza il regime sopra descritto, l’HbA1c è migliorata dal 13,3% all’8,4% (dopo 17 mesi) e poi al 6,9% (dopo ulteriori 19 mesi), e l’ICARS è diminuito da 85 a 71, con un miglioramento di 14 punti2).
QQuale trattamento è appropriato per il diabete associato all'atassia di Friedreich?
A
A causa della disfunzione mitocondriale sottostante, metformina e tiazolidinedioni devono essere evitati2). Sono raccomandati gli inibitori della DPP-4 (come sitagliptin) e gli analoghi del GLP-1. Terapie adiuvanti mirate ai mitocondri, come L-carnitina, CoQ10 e vitamina E, possono essere utili2).
6. Fisiopatologia e meccanismo dettagliato della malattia
La frataxina è una proteina localizzata nella membrana interna mitocondriale. È coinvolta nel metabolismo del ferro (immagazzinamento del ferro e assemblaggio dei cluster ferro-zolfo) ed è essenziale per il normale funzionamento della catena respiratoria mitocondriale1).
L’espansione delle ripetizioni GAA causa un silenziamento trascrizionale (transcriptional silencing) e riduce i livelli di mRNA di FXN1). La carenza di frataxina porta a quanto segue.
Accumulo di ferro mitocondriale: a causa di un’alterata elaborazione del ferro.
Ridotta produzione di ATP: disfunzione dei complessi della catena respiratoria2).
Aumento delle ROS: aumento dello stress ossidativo1)2).
Ganglio spinale : piccolo e atrofico, la radice posteriore è sottile e di colore grigio.
Midollo spinale: riduzione del diametro per tutta la lunghezza, particolarmente marcata nella regione toracica. Degenerazione progressiva dei tratti corticospinale, spinocerebellare e dei cordoni posteriori.
La carenza di frataxina provoca variazioni dei livelli di ferro intracellulare, rendendo le cellule gangliari retiniche (RGC) vulnerabili allo stress ossidativo. Fino al 30% dei pazienti con FA presenta segni oftalmici. Sono danneggiati sia i circuiti del tronco encefalico-cervelletto che il nervo ottico. La riduzione dello spessore della RNFL è direttamente correlata a una diminuzione dell’acuità visiva e della sensibilità al contrasto, e i difetti del campo visivo iniziali, che progrediscono concentricamente dalla periferia, sono associati alla perdita di RNFL documentata dall’OCT.
La cardiomiopatia ipertrofica aumenta il peso del cuore. Il tessuto miocardico può presentare macroscopicamente un aspetto «marmorizzato» (marble-like).
La disfunzione mitocondriale è la causa alla base del diabete associato a FRDA2).
Disfunzione delle cellule β : La ridotta produzione di ATP diminuisce la secrezione di insulina, portando infine alla perdita delle cellule β.
Alterata secrezione delle cellule α : Iperglucagonemia paradossa in iperglicemia e risposta glucagonica insufficiente in ipoglicemia2).
Meccanismi di resistenza all’insulina : Ridotta captazione del glucosio a causa di un’alterata fosforilazione ossidativa nel muscolo scheletrico, accumulo ectopico di grasso nel fegato e nei muscoli a causa di un alterato metabolismo lipidico, alterata segnalazione insulinica a causa di infiammazione cronica e stress ossidativo, anomala produzione epatica di glucosio a causa di neuropatia autonomica, e deterioramento metabolico secondario dovuto a inattività fisica e atrofia muscolare correlate all’atassia2).
I portatori di delezione intragenica tendono a presentare esordio più precoce, progressione rapida e cardiomiopatia grave rispetto all’espansione biallelica1). Se la delezione rimuove il codone di inizio, la produzione proteica viene completamente abolita1).
QPerché l'atassia di Friedreich causa sintomi oculari?
A
Il metabolismo anomalo del ferro e lo stress ossidativo dovuti alla carenza di frataxina danneggiano le cellule gangliari della retina. La riduzione dello spessore della RNFL è direttamente correlata a una diminuzione dell’acuità visiva e della sensibilità al contrasto, causando un difetto del campo visivo che progredisce concentricamente dalla periferia. Inoltre, il danno ai circuiti del tronco encefalico e del cervelletto provoca anomalie dei movimenti oculari come nistagmo, disturbi dei movimenti di inseguimento e disturbi della misurazione delle saccadi.
7. Ricerche recenti e prospettive future (rapporti in fase di ricerca)
Lo studio clinico di Cooper et al. (2008) ha riportato un miglioramento significativo del punteggio ICARS nell’arco di due anni con la terapia combinata di CoQ10 e vitamina E2).
Lo studio crossover randomizzato controllato con placebo di Schöls et al. (2005) ha mostrato che la somministrazione di L-carnitina migliorava significativamente la produzione di ATP mitocondriale2).
Sureshkumar et al. (2025) hanno trattato una donna di 32 anni con diabete associato ad atassia di Friedreich con una combinazione di terapia insulinica, sitagliptin, L-carnitina, CoQ10, vitamina E, vitamine neurotrope e imeglimina2). L’HbA1c è migliorata dal 13,3% all’8,4% dopo 17 mesi e ulteriormente al 6,9% dopo 19 mesi, e l’ICARS è sceso da 85 a 71, con un miglioramento di 14 punti (HOMA2-IR: 4,5→1,2; HOMA2-%B: 5→60; MAGE: 120→70 mg/dL; CV: 43%→34,9%). Mentre la storia naturale secondo SARA prevede un peggioramento di circa 2,31 punti in tre anni, è stata osservata una deviazione positiva di circa 16 punti. Questo è il primo caso riportato di stabilizzazione glicemica a lungo termine e miglioramento dell’atassia ottenuti simultaneamente.
Si ritiene che l’imeglimina agisca migliorando l’attività della catena respiratoria, riducendo lo stress ossidativo e promuovendo la sintesi di ATP/NAD+2).
Terapia con vettori AAV : Lo studio clinico AAVrh.10hFXN, volto a introdurre il gene FXN normale nel cuore e nel sistema nervoso, è in corso e ha mostrato un miglioramento precoce dell’espressione di FXN e dei marcatori di malattia (Munoz-Zuluaga et al. 2023)2).
CRISPR-Cas9 : mirato alla rimozione delle sequenze ripetute GAA, sono in corso studi su cellule e modelli murini YG8R (Ouellet et al. 2017)2).
Sfida : È necessario controllare la risposta immunitaria e stabilire un metodo sicuro di somministrazione genica2).
Miglioramento dell’accuratezza della diagnosi genetica
Aguilera et al. (2023) hanno identificato una nuova delezione intragenica che include la regione 5’UTR e gli esoni 1-2 del gene FXN analizzando campioni parentali di un paziente con presunta espansione biallelica 1). Finora in letteratura sono state riportate solo 10 delezioni intrageniche, ma si ipotizza che possano essere più frequenti. La standardizzazione delle analisi genetiche complementari mediante MLPA e dei test parentali è considerata una sfida futura.
La segnalazione del primo caso confermato in Africa occidentale evidenzia la necessità di studi di coorte su larga scala in diversi contesti etnici e geografici3). L’identificazione di varianti modificanti la malattia potrebbe rappresentare futuri bersagli terapeutici.
Aguilera C, Esteve-Garcia A, Casasnovas C, et al. Novel intragenic deletion within the FXN gene in a patient with typical phenotype of Friedreich ataxia: may be more prevalent than we think? BMC Med Genomics. 2023;16:312.
Sureshkumar P, Kumar SS, Cheriyan J, Masood A. Friedreich Ataxia and Related Diabetes: Therapeutic Approach Targeting Mitochondrial Dysfunction. JCEM Case Rep. 2025;3:luaf215.
Cissé CAK, Cissé L, Ba HO, et al. Friedreich ataxia in a family from Mali, West Africa. Clin Case Rep. 2021;9:e04065.
Copia il testo dell'articolo e incollalo nell'assistente IA che preferisci.
Articolo copiato negli appunti
Apri un assistente IA qui sotto e incolla il testo copiato nella chat.