Friedreich Ataxia (FRDA) is a progressive hereditary disease that affects the central and peripheral nervous systems. It is considered the most common autosomal recessive hereditary ataxia in the Caucasian population.
In 1863, Nikolaus Friedreich reported juvenile-onset ataxia, scoliosis, and familial cardiac degeneration. Later, Pierre Marie distinguished FRDA from other ataxias, establishing the disease concept.
Epidemiologically, there are large regional differences, with prevalence reported from 1:20,000 to 1:750,000. In Europeans, it is about 1:21,0001), and worldwide it is estimated at 1:40,0002) to 1:50,0003). The carrier frequency is about 1 in 701). It is more common in southern France, northern Spain, and Ireland, and less common in Scandinavia and Russia. It is even rarer in sub-Saharan Africa and the Eastern Hemisphere, but genetically confirmed cases have been reported in West Africa (in a consanguineous Tuareg family in Mali)3).
The inheritance pattern is autosomal recessive, and the incidence is equal in males and females. The typical age of onset is an average of 15.5 years, with most cases occurring before age 25. Onset is most common between 8 and 15 years of age2). The average life expectancy is 39 years, and the main cause of death is cardiomyopathy2).
QHow common is Friedreich Ataxia?
A
Friedreich ataxia is the most common hereditary ataxia in the Caucasian population, with prevalence ranging from 1:20,000 to 1:750,000 depending on the region. In Europe, it affects approximately 1 in 21,000 people1), and worldwide it is estimated to affect 1 in 40,000 to 50,000 people2)3). The carrier frequency is about 1 in 701).
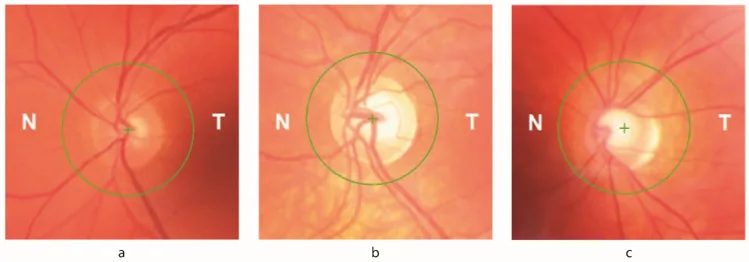
Petya Bogdanova-Mihaylova, Helena Maria Plapp, Hongying Chen et al. Longitudinal Assessment Using Optical Coherence Tomography in Patients with Friedreich’s Ataxia. Tomography. 2021 Dec 8; 7(4):915. Figure 1. PMCID: PMC8706975. License: CC BY.
Figure showing the main symptoms of Friedreich ataxia
Gait disturbance: The most common initial symptom. It begins with multidirectional unsteady gait.
Clumsiness and tremor of the upper limbs: Some cases present with hand tremor as the initial symptom3).
Dysarthria: Often appears within 10 to 15 years after onset.
Sensory symptoms: Loss of deep sensation (proprioception) occurs.
Visual symptoms: Some patients experience decreased visual acuity and contrast sensitivity, but most lack visual subjective symptoms in the early stages.
The progression of motor symptoms varies among individuals, but on average, patients become unable to walk independently about 8 years after the initial symptoms, and require a wheelchair within 11 to 15 years. In cases with longer GAA repeats, wheelchair dependence has been reported within 5 years 3).
Clinical Findings (Findings Confirmed by Physician Examination)
Ataxic gait: Unstable gait in all directions. Accompanied by dysmetria, hypotonia, dysdiadochokinesia, and impaired coordination of movement timing 1).
Loss of deep tendon reflexes: Loss of patellar and Achilles tendon reflexes is a mandatory item in the Harding diagnostic criteria. Loss of upper limb reflexes is also frequently associated. Some cases present with hyperreflexia, spasticity, and scissor gait 3).
Babinski sign: Appears as an extensor plantar response.
Skeletal deformities: Scoliosis (appears early), pes cavus, lumbar lordosis3).
Systemic complications: Hypertrophic cardiomyopathy (up to 63%), diabetes mellitus (5–40%)2).
Neuro-ophthalmologic findings
Nystagmus: Due to cerebellar/brainstem circuit dysfunction. Includes gaze-evoked nystagmus.
Square-wave jerks: Involuntary saccadic intrusions during fixation.
Saccadic dysmetria: Hypermetric or hypometric saccades.
Optic atrophy: Detected on fundus examination. Ophthalmic signs appear in up to 30% of patients.
RNFL thinning: detected by OCT. Directly correlates with decreased visual acuity and contrast sensitivity.
Visual field defects: early concentric progression from the periphery. Associated with RNFL loss recorded by OCT.
QDo ocular symptoms always appear?
A
In FRDA, up to 30% of patients develop ocular signs, but many patients lack early visual symptoms. Optic atrophy, RNFL thinning, and eye movement disorders are detected by objective tests and directly correlate with decreased visual acuity and contrast sensitivity. Regular ophthalmic examinations are recommended.
FRDA is caused by an expansion of GAA trinucleotide repeats in intron 1 of the FXN (frataxin) gene on chromosome 9. 96% of cases are homozygous for biallelic pathogenic expansions1). The remaining cases are compound heterozygotes with a GAA expansion and a point mutation, or a GAA expansion and an intragenic/whole-gene deletion1).
The relationship between GAA repeat number and disease is shown below.
Classification
GAA repeat number
Normal
5–33 repeats
Intermediate (carrier equivalent)
34–65 repeats
Pathogenic
66 repeats or more (90 repeats or more in other reports) 1)3)
The longer the GAA repeat, the earlier the age of onset and the more severe the disease tends to be. In a case with extremely long allele repeats of 999/766, onset occurred at age 11 and a wheelchair was needed within 5 years (faster than the typical 10 years) 3). Interruptions within the repeat sequence may delay the age of onset 1).
Compound heterozygotes with an intragenic deletion plus a GAA expansion are thought to have earlier onset, faster progression, and more severe cardiomyopathy than biallelic expansions, although cases with a typical course have also been reported 1).
Consanguineous marriage increases the risk of onset due to the autosomal recessive nature of the disease 2)3). The condition is more frequent in populations of European ancestry.
QIf genetic testing shows a homozygous result, is it possible that the patient is actually a compound heterozygote?
A
PCR fragment analysis and TP-PCR cannot detect intragenic deletions, so an apparent biallelic expansion may actually be a GAA expansion plus an intragenic deletion 1). Additional testing with MLPA (Multiplex Ligation-dependent Probe Amplification) and parental sample testing are essential for accurate genetic counseling.
Harding’s criteria are used for clinical diagnosis.
Category
Main Items
Essential Items
Onset before age 25, progressive gait and limb ataxia, loss of patellar and Achilles tendon reflexes, axonal neuropathy, dysarthria (within 5 years of onset)
Additional findings (≥66%)
Scoliosis, lower limb pyramidal weakness, absent upper limb reflexes, loss of large-fiber sensation, abnormal ECG
PCR + TP-PCR: Amplify the GAA repeat in intron 1 to detect expanded alleles.
MLPA (Multiplex Ligation-dependent Probe Amplification): Required for detecting deletions/duplications not identified by fragment analysis or TP-PCR1). Added to rule out apparent biallelic expansion being a compound heterozygous state.
Southern blot analysis: Combined with PCR enables diagnosis with >99% accuracy3).
Electrooculography (EOG): Used for qualitative and quantitative analysis of eye movements. Evaluates visually guided saccades (latency and amplitude), pursuit (gain at different target velocities), VOR (gain at different head angular velocities), and fixation (nystagmus waveform analysis, square-wave latency).
Optical coherence tomography (OCT): Quantitatively measures retinal nerve fiber layer (RNFL) thickness. Can confirm correlation with decreased visual acuity and contrast sensitivity.
Visual field testing: Evaluates concentric peripheral visual field loss progressing inward.
Electrocardiogram and echocardiography : Assess cardiomyopathy, left ventricular wall thickness, ejection fraction, and diastolic function2). Regular monitoring is required.
Blood glucose-related tests2): Fasting blood glucose, HbA1c, C-peptide, fasting insulin, pancreatic islet autoantibodies (GAD65, IA-2, ZnT8), HOMA2-IR, and HOMA2-%B are used for early detection and classification of diabetes.
Clinical rating scales: FARS (Friedreich Ataxia Rating Scale), ICARS, and SARA are used to quantitatively assess neurological symptoms2). SARA shows a natural progression of 0.77 points per year (SE 0.06)2).
Brain and spinal cord MRI: Evaluates cerebellar atrophy (which may appear 3 years after onset3)) and spinal cord degeneration.
Currently, there is no curative treatment, and the mainstay of therapy is symptomatic management and complication control through multidisciplinary collaboration.
Scoliosis management: Bracing for mild to moderate cases; surgery considered for severe cases.
Pes cavus management: Botulinum toxin injection into the gastrocnemius muscle and Achilles tendon stretching to improve mobility.
Rehabilitation: Physical therapy, occupational therapy, and speech therapy to maintain function.
Ophthalmology
Low vision care: Use of magnifiers, lighting adjustments, and lifestyle guidance for patients with visual symptoms due to optic atrophy or retinal atrophy.
Symptomatic treatment for eye movement disorders: Prism glasses (adding equal prism power to both eyes in the direction of worsening gaze) for gaze-evoked nystagmus. GABA_B agonists are administered for vertical nystagmus, periodic alternating nystagmus, and saccadic intrusions.
Prescription example (GABA_B agonist): Gabaron tablets (5 mg) 3–6 tablets, divided into 1–3 doses.
Diabetes associated with FRDA is due to underlying mitochondrial dysfunction, so caution is needed in drug selection2).
Medications to avoid: Metformin and thiazolidinediones should be avoided because they inhibit mitochondrial complex I2). Sulfonylureas carry risks of beta-cell stress and hypoglycemia.
Recommended glucose-lowering drugs: DPP-4 inhibitors (e.g., sitagliptin 100 mg/day) and GLP-1 analogs are preferred2). Imeglimin (500 mg twice daily) may be useful as a novel glucose-lowering agent targeting mitochondrial function2).
Insulin therapy: Start at approximately 0.5 U/kg/day2).
Mitochondria-targeted adjunctive therapy: A combination of L-carnitine 500 mg/day, CoQ10 100 mg/day, and vitamin E 400 IU/day has been reported2).
In a case report using the above regimen, HbA1c improved from 13.3% to 8.4% (after 17 months) and further to 6.9% (after an additional 19 months), and ICARS improved by 14 points from 85 to 712).
QWhat treatment is appropriate for diabetes associated with Friedreich's ataxia?
A
Because mitochondrial dysfunction underlies the condition, metformin and thiazolidinediones should be avoided 2). DPP-4 inhibitors (e.g., sitagliptin) and GLP-1 analogs are recommended. Mitochondria-targeted adjuvant therapies such as L-carnitine, CoQ10, and vitamin E may also be beneficial 2).
Frataxin is a protein localized to the inner mitochondrial membrane. It is involved in iron metabolism (iron storage and assembly of iron-sulfur clusters) and is essential for normal function of the mitochondrial respiratory chain 1).
Expansion of GAA repeats causes transcriptional silencing, reducing FXN mRNA levels 1). Frataxin deficiency leads to the following:
Mitochondrial iron accumulation: Due to impaired iron handling.
Decreased ATP production: Due to dysfunction of respiratory chain complexes2).
Dorsal root ganglia: Small and atrophic; dorsal roots are thin and gray.
Spinal cord: Reduced diameter throughout, especially in the thoracic region. Progressive degeneration of the corticospinal tracts, spinocerebellar tracts, and posterior columns occurs.
Frataxin deficiency alters intracellular iron levels, making retinal ganglion cells (RGCs) vulnerable to oxidative stress. Up to 30% of FA patients develop ophthalmic signs. Both the brainstem-cerebellar circuits and the optic nerve are affected. Reduction in RNFL thickness directly correlates with decreased visual acuity and contrast sensitivity, and early concentric peripheral visual field defects are associated with RNFL loss documented by OCT.
Individuals with intragenic deletions tend to have earlier onset, more rapid progression, and more severe cardiomyopathy than those with biallelic expansions 1). If the deletion removes the start codon, protein production is completely abolished 1).
QWhy do eye symptoms occur in Friedreich's ataxia?
A
Iron metabolism abnormalities and oxidative stress due to frataxin deficiency damage retinal ganglion cells. Reduction in RNFL thickness directly correlates with decreased visual acuity and contrast sensitivity, leading to visual field defects that progress concentrically from the periphery. Additionally, impairment of brainstem and cerebellar circuits causes eye movement abnormalities such as nystagmus, impaired smooth pursuit, and saccadic dysmetria.
7. Latest Research and Future Perspectives (Investigational Reports)
A clinical trial by Cooper et al. (2008) reported that combination therapy with CoQ10 and vitamin E significantly improved ICARS scores over a 2-year period2).
A randomized placebo-controlled crossover trial by Schöls et al. (2005) showed that L-carnitine administration significantly improved mitochondrial ATP production2).
Sureshkumar et al. (2025) administered a combination of insulin therapy, sitagliptin, L-carnitine, CoQ10, vitamin E, neurotropic vitamins, and imeglimin to a 32-year-old woman with FA-related diabetes2). HbA1c improved from 13.3% to 8.4% after 17 months and further to 6.9% after 19 months, and ICARS improved by 14 points from 85 to 71 (HOMA2-IR: 4.5→1.2, HOMA2-%B: 5→60, MAGE: 120→70 mg/dL, CV: 43%→34.9%). While the natural history by SARA predicts a worsening of approximately 2.31 points over 3 years, this case showed a positive deviation of about 16 points. This is considered the first reported case of simultaneous achievement of long-term glycemic stabilization and ataxia improvement.
Imeglimin is thought to act through improvement of respiratory chain activity, reduction of oxidative stress, and promotion of ATP/NAD+ synthesis2).
AAV vector therapy: Clinical trials of AAVrh.10hFXN are ongoing to deliver the normal FXN gene to the heart and nervous system, with early improvements in FXN expression and disease markers reported (Munoz-Zuluaga et al. 2023)2).
CRISPR-Cas9: Aimed at removing GAA repeat expansions, studies are progressing in YG8R-derived cells and mouse models (Ouellet et al. 2017)2).
Challenges: Control of immune responses and establishment of safe gene delivery methods are needed2).
Aguilera et al. (2023) identified a novel intragenic deletion involving the 5’UTR and exons 1–2 of the FXN gene by testing parental samples from a patient presumed to have biallelic expansions1). Although only 10 intragenic deletions have been reported in the literature, it is suggested that they may actually be more frequent. Standardization of complementary genetic analysis using MLPA and parental sample testing are future challenges.
The first confirmed case in West Africa highlights the need for large-scale cohort studies across diverse ethnic and geographic backgrounds3). Identification of disease-modifying variants may serve as future therapeutic targets.
Aguilera C, Esteve-Garcia A, Casasnovas C, et al. Novel intragenic deletion within the FXN gene in a patient with typical phenotype of Friedreich ataxia: may be more prevalent than we think? BMC Med Genomics. 2023;16:312.
Sureshkumar P, Kumar SS, Cheriyan J, Masood A. Friedreich Ataxia and Related Diabetes: Therapeutic Approach Targeting Mitochondrial Dysfunction. JCEM Case Rep. 2025;3:luaf215.
Cissé CAK, Cissé L, Ba HO, et al. Friedreich ataxia in a family from Mali, West Africa. Clin Case Rep. 2021;9:e04065.
Copy the article text and paste it into your preferred AI assistant.
Article copied to clipboard
Open an AI assistant below and paste the copied text into the chat box.