โรคฟรีดริชส์อะแท็กเซีย (FRDA) เป็นโรคอะแท็กเซียทางพันธุกรรมแบบถอย autosomal ที่พบบ่อยที่สุดในประชากรผิวขาว
สาเหตุเกิดจากการขยายตัวของลำดับเบส GAA ซ้ำแบบ homozygous ในอินตรอน 1 ของยีน FXN บนโครโมโซมคู่ที่ 9 ซึ่งพบใน 96% ของผู้ป่วย
อายุที่เริ่มมีอาการโดยทั่วไปคือ 8–15 ปี และอาการแรกที่พบบ่อยที่สุดคือความผิดปกติในการเดิน
โรคนี้มีภาวะแทรกซ้อนหลายระบบ ได้แก่ โรคกล้ามเนื้อหัวใจ (สูงสุด 63%) เบาหวาน (5–40%) และฝ่อของเส้นประสาทตา
ผู้ป่วยสูงสุด 30% มีอาการทางตา เช่น ฝ่อของเส้นประสาทตา อาตา และความผิดปกติของการเคลื่อนไหวลูกตา
ยังไม่มีการรักษาที่หายขาด การรักษาหลักคือการรักษาตามอาการและการจัดการภาวะแทรกซ้อนโดยสหสาขาวิชาชีพ
อายุขัยเฉลี่ยคือ 39 ปี และสาเหตุการเสียชีวิตหลักคือโรคกล้ามเนื้อหัวใจ
โรคฟรีดไรค์อะแท็กเซีย (Friedreich Ataxia; FRDA) เป็นโรคทางพันธุกรรมที่ลุกลามซึ่งส่งผลต่อระบบประสาทส่วนกลางและระบบประสาทส่วนปลาย ถือเป็นโรคอะแท็กเซียชนิดถ่ายทอดทางพันธุกรรมแบบด้อยที่พบบ่อยที่สุดในกลุ่มคนผิวขาว
ในปี ค.ศ. 1863 นิโคเลาส์ ฟรีดไรค์รายงานอาการอะแท็กเซีย กระดูกสันหลังคด และโรคหัวใจเสื่อมในครอบครัวที่เริ่มมีอาการตั้งแต่อายุน้อย ต่อมาปิแอร์ มารีได้แยก FRDA ออกจากโรคอะแท็กเซียชนิดอื่น ทำให้แนวคิดของโรคนี้ได้รับการยอมรับ
ทางระบาดวิทยา ความชุกของโรคแตกต่างกันมากในแต่ละภูมิภาค โดยรายงานว่าอยู่ระหว่าง 1:20,000 ถึง 1:750,000 ในชาวยุโรปประมาณ 1:21,0001) และทั่วโลกประมาณ 1:40,0002) ถึง 1:50,0003) ความถี่ของพาหะประมาณ 1/701) พบความถี่สูงในฝรั่งเศสตอนใต้ สเปนตอนเหนือ และไอร์แลนด์ แต่ต่ำในสแกนดิเนเวียและรัสเซีย ในแอฟริกาใต้สะฮาราและซีกโลกตะวันออกพบน้อยกว่า แต่ก็มีรายงานการยืนยันทางพันธุกรรมในแอฟริกาตะวันตก (ในครอบครัวชาวทูอาเร็กที่มีการแต่งงานในเครือญาติในประเทศมาลี)3)
รูปแบบการถ่ายทอดทางพันธุกรรมเป็นแบบด้อย อัตราการเกิดโรคในเพศชายและหญิงเท่ากัน อายุที่เริ่มมีอาการโดยทั่วไปเฉลี่ย 15.5 ปี ส่วนใหญ่เริ่มมีอาการก่อนอายุ 25 ปี มักเริ่มมีอาการระหว่างอายุ 8-15 ปี2) อายุขัยเฉลี่ย 39 ปี สาเหตุการเสียชีวิตหลักคือโรคกล้ามเนื้อหัวใจ2)
Q
โรคฟรีดไรค์อะแท็กเซียเกิดขึ้นบ่อยแค่ไหน?
A
เป็นโรคทางพันธุกรรมที่ทำให้เกิดการเคลื่อนไหวผิดปกติที่พบบ่อยที่สุดในกลุ่มคนผิวขาว โดยความชุกแตกต่างกันไปตามภูมิภาค ตั้งแต่ 1:20,000 ถึง 1:750,000 ในยุโรปประมาณ 1 ใน 21,000 คน1) และทั่วโลกประมาณ 1 ใน 40,000 ถึง 50,000 คน2) 3) ความถี่ของพาหะประมาณ 1 ใน 70 คน1)
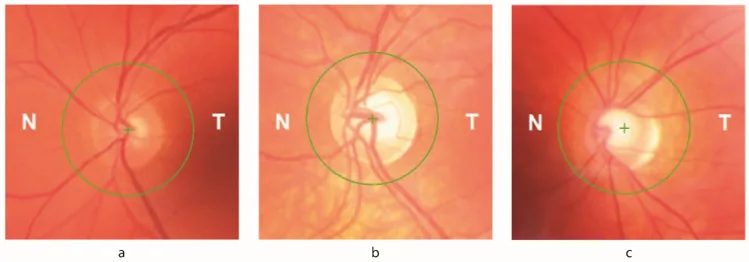
Petya Bogdanova-Mihaylova, Helena Maria Plapp, Hongying Chen et al. Longitudinal Assessment Using Optical Coherence Tomography in Patients with Friedreich’s Ataxia. Tomography. 2021 Dec 8; 7(4):915. Figure 1. PMCID: PMC8706975. License: CC BY.
ภาพแสดงลักษณะของโรค Friedreich ataxia
ความผิดปกติในการเดิน : อาการเริ่มแรกที่พบบ่อยที่สุด เริ่มจากการเดินไม่มั่นคงในทุกทิศทางความเก้กังของแขนและมือสั่น : บางรายเริ่มต้นด้วยอาการมือสั่น3) การพูดไม่ชัด : มักเกิดขึ้นภายใน 10-15 ปีหลังจากเริ่มมีอาการอาการทางประสาทสัมผัส : สูญเสียการรับรู้ความรู้สึกลึก (proprioception)อาการทางตา : ผู้ป่วยบางรายอาจมีสายตาลดลงหรือความไวต่อความคมชัดลดลง แต่ผู้ป่วยส่วนใหญ่ในระยะแรกไม่มีอาการทางตาที่ชัดเจน
การดำเนินของอาการทางการเคลื่อนไหวแตกต่างกันในแต่ละบุคคล แต่โดยเฉลี่ยประมาณ 8 ปีหลังจากเริ่มมีอาการแรก ผู้ป่วยจะไม่สามารถเดินได้เอง และต้องใช้วีลแชร์ภายใน 11–15 ปี ในกรณีที่มีจำนวนซ้ำ GAA มาก อาจต้องใช้วีลแชร์ภายใน 5 ปี 3)
อาการแสดงทางระบบประสาททั่วร่างกาย
การเดินแบบ ataxic : การเดินไม่มั่นคงในทุกทิศทาง ร่วมกับ dysmetria, hypotonia, dysdiadochokinesia และ impaired check reflex 1)
การหายไปของ deep tendon reflex : การหายไปของ patellar reflex และ Achilles reflex เป็นข้อบังคับในเกณฑ์วินิจฉัยของ Harding มักพบร่วมกับการหายไปของรีเฟล็กซ์แขนด้วย ในบางรายอาจมีรีเฟล็กซ์เกิน, spasticity หรือ scissor gait 3)
Babinski sign : ปรากฏเป็น plantar response แบบ extension
ความผิดปกติของโครงกระดูก : กระดูกสันหลังคด (ปรากฏตั้งแต่ระยะแรก), เท้าแหว่ง (pes cavus), กระดูกสันหลังส่วนเอวโค้งงอ3)
ภาวะแทรกซ้อนทั่วร่างกาย : กล้ามเนื้อหัวใจหนาตัวผิดปกติ (สูงสุด 63%), เบาหวาน (5–40%)2)
อาการทางประสาทจักษุวิทยา
อาตา (nystagmus)อาตา ที่ขึ้นกับตำแหน่งตา
square-wave jerks : การเคลื่อนไหวของลูกตาแบบกระตุกโดยไม่สมัครใจที่แทรกระหว่างการจ้องมอง
ความผิดปกติของการเคลื่อนไหวตามเป้าหมาย : การติดตามเป้าหมายเป็นขั้นบันได (ความผิดปกติของการไล่ตาม)
saccadic dysmetria : การกระโดดของลูกตาที่มากเกินไปหรือน้อยเกินไป
ฝ่อของเส้นประสาทตา
ความหนาของชั้น RNFL ลดลง : ตรวจพบโดย OCT สัมพันธ์โดยตรงกับการมองเห็น และความไวต่อคอนทราสต์ที่ลดลง
ข้อบกพร่องของลานสายตา : ข้อบกพร่องของลานสายตาระยะแรกที่ลุกลามเป็นวงกลมจากบริเวณรอบนอก สัมพันธ์กับการสูญเสีย RNFL ที่บันทึกโดย OCT
Q
อาการทางตาจะต้องเกิดขึ้นเสมอหรือไม่?
A
ใน FRDA ผู้ป่วยสูงสุด 30% มีอาการแสดงทางตา แต่ผู้ป่วยจำนวนมากไม่มีอาการทางสายตาในระยะแรก ฝ่อของเส้นประสาทตา ความหนาของ RNFL ลดลง และความผิดปกติของการเคลื่อนไหวของลูกตาสามารถตรวจพบได้โดยการตรวจตามวัตถุประสงค์ และสัมพันธ์โดยตรงกับการมองเห็น และความไวต่อคอนทราสต์ที่ลดลง แนะนำให้ตรวจตาเป็นประจำ
สาเหตุของ FRDA คือการขยายตัวของลำดับเบส GAA ซ้ำสามเบสในอินตรอน 1 ของยีน FXN (ฟราทาซิน) บนโครโมโซมคู่ที่ 9 ผู้ป่วย 96% เป็นโฮโมไซกัสสำหรับการขยายตัวที่ก่อโรคแบบ biallelic 1) ส่วนที่เหลือเป็นคอมพาวด์เฮเทอโรไซกัสระหว่างการขยาย GAA กับการกลายพันธุ์แบบจุด หรือการขยาย GAA กับการลบภายในยีน/ทั้งยีน 1)
ความสัมพันธ์ระหว่างจำนวนซ้ำของ GAA กับโรคแสดงไว้ด้านล่าง
การจำแนก จำนวนซ้ำ GAA ปกติ 5–33 ครั้ง ระดับกลาง (เทียบเท่าพาหะ) 34–65 ครั้ง ก่อโรค 66 ครั้งขึ้นไป (รายงานอื่นระบุ 90 ครั้งขึ้นไป) 1) 3)
จำนวนซ้ำ GAA ที่ยาวขึ้นสัมพันธ์กับอายุที่เริ่มมีอาการเร็วขึ้นและอาการรุนแรงขึ้น ในกรณีที่มีจำนวนซ้ำของอัลลีลยาวมากถึง 999/766 ผู้ป่วยเริ่มมีอาการเมื่ออายุ 11 ปี และต้องใช้วีลแชร์ภายใน 5 ปี (เร็วกว่าปกติซึ่งประมาณ 10 ปี) 3) การขัดจังหวะ (interruption) ภายในลำดับซ้ำอาจทำให้อายุที่เริ่มมีอาการช้าลง 1)
ผู้ที่มีการกลายพันธุ์แบบ compound heterozygous (intragenic deletion ร่วมกับ GAA expansion) มักมีแนวโน้มที่จะเริ่มมีอาการเร็วขึ้น ดำเนินโรคเร็วขึ้น และมีภาวะกล้ามเนื้อหัวใจผิดปกติรุนแรงกว่าเมื่อเทียบกับผู้ที่มี biallelic expansion แต่อย่างไรก็ตาม ก็มีรายงานผู้ป่วยที่มีอาการดำเนินไปในลักษณะปกติเช่นกัน 1)
การแต่งงานในเครือญาติเพิ่มความเสี่ยงต่อการเกิดโรคเนื่องจากลักษณะการถ่ายทอดทางพันธุกรรมแบบ autosomal recessive 2) 3) โรคนี้พบได้บ่อยในประชากรที่มีบรรพบุรุษเป็นชาวยุโรป
การให้คำปรึกษาทางพันธุกรรม
FRDA เป็นโรคที่ถ่ายทอดทางพันธุกรรมแบบ autosomal recessive หากพ่อและแม่เป็นพาหะ โอกาสที่ลูกจะเป็นโรคคือ 25% แนะนำให้ปรึกษานักพันธุศาสตร์เมื่อเข้ารับการตรวจวินิจฉัยทางพันธุกรรม
Q
หากผลการตรวจทางพันธุกรรมระบุว่าเป็น homozygous มีความเป็นไปได้หรือไม่ที่จะเป็น compound heterozygous?
A
การวิเคราะห์ชิ้นส่วน PCR และ TP-PCR ไม่สามารถตรวจพบการขาดหายภายในยีนได้ ดังนั้น biallelic expansion ที่เห็นอาจเป็น GAA expansion ร่วมกับการขาดหายภายในยีน (intragenic deletion) 1) การตรวจเพิ่มเติมด้วย MLPA (Multiplex Ligation-dependent Probe Amplification) และการตรวจตัวอย่างจากพ่อแม่เป็นสิ่งจำเป็นสำหรับการให้คำปรึกษาทางพันธุกรรม ที่แม่นยำ
การวินิจฉัยทางคลินิกใช้เกณฑ์ของ Harding
หมวดหมู่ รายการหลัก ข้อบังคับ เริ่มมีอาการก่อนอายุ 25 ปี, การเดินผิดปกติและการเคลื่อนไหวไม่ประสานกันที่แขนขาแบบค่อยเป็นค่อยไป, การหายไปของรีเฟล็กซ์เอ็นสะบ้าและเอ็นร้อยหวาย, ลักษณะการเสื่อมของแอกซอน, และอาการพูดไม่ชัด (หลังจากเริ่มมีอาการ 5 ปี) อาการเพิ่มเติม (มากกว่า 66%) กระดูกสันหลังคด กล้ามเนื้อขาอ่อนแรงจากทางเดินคอร์ติโคสไปนัล ปลายแขนขาดรีเฟล็กซ์ สูญเสียความรู้สึกของเส้นใยประสาทขนาดใหญ่ คลื่นไฟฟ้าหัวใจผิดปกติ อื่นๆ (น้อยกว่า 50%) อาตา จอประสาทตา ฝ่อ หูหนวก กล้ามเนื้อส่วนปลายลีบ เท้าโก่ง เบาหวาน
PCR + TP-PCR : เพิ่มจำนวนลำดับซ้ำ GAA ในอินทรอน 1 เพื่อตรวจหาอัลลีลที่ยืดยาวMLPA (Multiplex Ligation-dependent Probe Amplification) : จำเป็นสำหรับการวินิจฉัยการขาดหายหรือการเพิ่มขึ้นของชิ้นส่วนดีเอ็นเอที่ไม่สามารถตรวจพบได้ด้วยการวิเคราะห์ชิ้นส่วนหรือ TP-PCR 1) เพิ่มเติมเพื่อแยกความเป็นไปได้ที่การขยายตัวแบบ biallelic ที่เห็นอาจเป็น compound heterozygousการวิเคราะห์ Southern blot : ร่วมกับ PCR สามารถวินิจฉัยได้แม่นยำมากกว่า 99% 3) การตรวจตัวอย่างจากพ่อแม่ : มีความจำเป็นสำหรับการให้คำปรึกษาทางพันธุกรรม ที่แม่นยำ1)
แสดงภาวะประสาทรับความรู้สึกเสื่อม โดยมีลักษณะเฉพาะคือการหายไปของศักย์ไฟฟ้าประสาทรับความรู้สึก (SNAP) รูปแบบของปลายประสาทรับความรู้สึกหลายเส้นเสื่อมแบบแอกซอนเป็นลักษณะเฉพาะ3)
การตรวจคลื่นไฟฟ้าตา (EOG ) : ใช้สำหรับการวิเคราะห์เชิงคุณภาพและเชิงปริมาณของการเคลื่อนไหวของลูกตา ประเมินการกลอกตาแบบซักเคดตามสิ่งเร้าทางสายตา (ระยะแฝงและแอมพลิจูด) การกลอกตาแบบเพอร์ซูท (อัตราขยายตามความเร็วเป้าหมายที่ต่างกัน) การตอบสนองต่อการหมุนศีรษะ (VOR) (อัตราขยายตามความเร็วเชิงมุมของศีรษะที่ต่างกัน) และการจ้องมอง (การวิเคราะห์รูปคลื่นอาตา ระยะแฝงของคลื่นสี่เหลี่ยม)เครื่องตรวจชั้นจอประสาทตา ด้วยแสง (OCT ) : วัดความหนาของชั้นใยประสาทจอตา (RNFL ) เชิงปริมาณ สามารถยืนยันความสัมพันธ์กับการลดลงของการมองเห็น และความไวต่อคอนทราสต์การตรวจลานสายตา การตรวจอวัยวะภายในลูกตา : ตรวจสอบการมีหรือไม่มีฝ่อของเส้นประสาทตา
เมื่อผู้ป่วยมีอาการหลักคือความผิดปกติของการเคลื่อนไหวของลูกตา จำเป็นต้องแยกโรคจากโรคต่อไปนี้
โรคสมองน้อยและไขสันหลังเสื่อมชนิดประปราย (SCD) : รวมถึงโรคระบบหลายส่วนฝ่อ (multiple system atrophy) บางส่วนโรคไคอารี (Chiari malformation) : มีลักษณะเด่นคืออาการตาพร่า จากการสั่นของลูกตาแบบลงล่าง (downbeat nystagmus)SCD ชนิดพันธุกรรม : เช่น SCA ชนิด 6, 31, 3โรคที่เกิดภายหลัง : รอยโรคที่สมองน้อยหรือก้านสมองจากการอักเสบ เนื้องอก หรือโรคหลอดเลือด
คลื่นไฟฟ้าหัวใจและ echocardiography : ประเมินภาวะกล้ามเนื้อหัวใจผิดปกติ ความหนาของผนังหัวใจห้องล่างซ้าย สัดส่วนการบีบตัวของหัวใจ และการทำงานของหัวใจในระยะคลายตัว2) จำเป็นต้องติดตามผลเป็นระยะการตรวจที่เกี่ยวข้องกับระดับน้ำตาลในเลือด 2) : ตรวจระดับน้ำตาลในเลือดขณะอดอาหาร HbA1c C-peptide ระดับอินซูลินขณะอดอาหาร แอนติบอดีต่อเซลล์ตับอ่อน (GAD65, IA-2, ZnT8) HOMA2-IR และ HOMA2-%B เพื่อการตรวจพบเบาหวานในระยะเริ่มต้นและการจำแนกชนิดของโรคมาตราวัดทางคลินิก : FARS (Friedreich Ataxia Rating Scale), ICARS, SARA สำหรับการประเมินอาการทางระบบประสาทในเชิงปริมาณ2) โดย SARA รายงานว่ามีการเสื่อมลงตามธรรมชาติ 0.77 คะแนนต่อปี (SE 0.06)2) MRI สมองและไขสันหลัง : ประเมินการฝ่อของสมองน้อย (มีรายงานพบหลังจากเริ่มมีอาการ 3 ปี3) ) และการเสื่อมของไขสันหลัง
ในปัจจุบันยังไม่มีการรักษาที่สามารถรักษาให้หายขาดได้ การรักษาหลักคือการรักษาตามอาการและการจัดการภาวะแทรกซ้อนโดยการทำงานร่วมกันของสหสาขาวิชาชีพ
อายุรศาสตร์ระบบประสาทและศัลยศาสตร์กระดูกและข้อ
การจัดการกระดูกสันหลังคด : กรณีเล็กน้อยถึงปานกลางใช้การใส่เครื่องดัดกระดูกสันหลัง กรณีรุนแรงพิจารณาการผ่าตัด
การจัดการเท้าแอ่น : ฉีดโบทูลินัมท็อกซินเข้ากล้ามเนื้อน่อง ยืดเอ็นร้อยหวายเพื่อเพิ่มความคล่องตัว
การฟื้นฟูสมรรถภาพ : กายภาพบำบัด กิจกรรมบำบัด และการบำบัดทางการพูดเพื่อรักษาการทำงาน
จักษุวิทยา
การดูแลผู้มีสายตาเลือนราง ฝ่อของเส้นประสาทตา และการฝ่อของจอประสาทตา ให้ใช้แว่นขยาย ปรับแสง และให้คำแนะนำการใช้ชีวิต
การรักษาตามอาการของความผิดปกติของการเคลื่อนไหวลูกตา : สำหรับอาตา ที่ขึ้นกับทิศทางการมอง ใช้แว่นปริซึม (เพิ่มปริซึมกำลังเท่ากันทั้งสองข้างในทิศทางที่ทำให้อาการแย่ลง) สำหรับอาตา แนวตั้ง อาตา แบบสลับทิศทางเป็นระยะ และการเคลื่อนไหวลูกตาแบบกระตุก ให้ใช้ยา GABA_B agonist
ตัวอย่างใบสั่งยา (GABA_B agonist) : Gabaron 5 มก. 3-6 เม็ด แบ่งให้วันละ 1-3 ครั้ง
โรคเบาหวานที่เกิดร่วมกับ FRDA มีสาเหตุจากความผิดปกติของไมโทคอนเดรีย ดังนั้นจึงต้องระมัดระวังในการเลือกใช้ยา2)
ยาที่ควรหลีกเลี่ยง : ควรหลีกเลี่ยงเมตฟอร์มินและไทอะโซลิดินไดโอนเนื่องจากยับยั้งไมโตคอนเดรียคอมเพล็กซ์ I 2) ซัลโฟนิลยูเรียมีความเสี่ยงต่อความเครียดของเซลล์เบต้าและภาวะน้ำตาลในเลือดต่ำยาลดน้ำตาลในเลือดที่แนะนำ : ควรใช้ DPP-4 inhibitors (เช่น sitagliptin 100 มก./วัน) และ GLP-1 analogs 2) อิเมกลิมิน (500 มก. วันละ 2 ครั้ง) อาจเป็นยาลดน้ำตาลในเลือดชนิดใหม่ที่มุ่งเป้าไปที่การทำงานของไมโตคอนเดรีย 2) การรักษาด้วยอินซูลิน : เริ่มต้นที่ประมาณ 0.5 ยูนิต/กก./วัน 2) การรักษาเสริมที่มุ่งเป้าไมโตคอนเดรีย : มีรายงานการใช้ L-carnitine 500 มก./วัน, CoQ10 100 มก./วัน และวิตามินอี 400 IU/วัน ร่วมกัน 2) วิตามินบำรุงระบบประสาท : B1 10 มก., B2 10 มก., B3 45 มก., B5 50 มก., B6 3 มก., B12 15 ไมโครกรัม/วัน 2)
ในรายงานผู้ป่วยที่ใช้สูตรการรักษาข้างต้น พบว่า HbA1c ลดลงจาก 13.3% เป็น 8.4% (หลัง 17 เดือน) และเป็น 6.9% (เพิ่มอีก 19 เดือน) และคะแนน ICARS ลดลง 14 คะแนนจาก 85 เป็น 71 2)
ไม่มีการรักษาให้หายขาด และอาการทางระบบประสาทจะดำเนินไปเรื่อยๆ
ในโรคเบาหวานที่เกิดร่วมกับ FRDA จำเป็นต้องเลือกใช้ยาที่แตกต่างจากเบาหวานชนิดที่ 2 ทั่วไป ควรหลีกเลี่ยงเมตฟอร์มินและไทอะโซลิดินไดโอนซึ่งยับยั้งไมโตคอนเดรียคอมเพล็กซ์ I 2)
โรคกล้ามเนื้อหัวใจเป็นสาเหตุหลักของการเสียชีวิต การประเมินหัวใจเป็นประจำ (คลื่นไฟฟ้าหัวใจและคลื่นเสียงสะท้อนหัวใจ) เป็นสิ่งจำเป็น
Q
การรักษาโรคเบาหวานที่เกิดร่วมกับโรคฟรีดไรช์แอทาเซียที่เหมาะสมคืออะไร?
A
เนื่องจากมีความผิดปกติของไมโตคอนเดรียเป็นพื้นฐาน จึงควรหลีกเลี่ยงเมตฟอร์มินและไทอะโซลิดินไดโอน 2) แนะนำให้ใช้ยา DPP-4 inhibitor (เช่น สิตากลิปติน) และ GLP-1 analog การรักษาเสริมที่มุ่งเป้าไปที่ไมโตคอนเดรีย เช่น แอล-คาร์นิทีน โคเอ็นไซม์คิวเท็น และวิตามินอี อาจมีประโยชน์ 2)
ฟราทาซินเป็นโปรตีนที่อยู่ที่เยื่อหุ้มชั้นในของไมโตคอนเดรีย มีบทบาทในการเผาผลาญธาตุเหล็ก (การเก็บธาตุเหล็กและการประกอบคลัสเตอร์เหล็ก-กำมะถัน) และจำเป็นต่อการทำงานปกติของห่วงโซ่การหายใจของไมโตคอนเดรีย 1)
การยืดตัวของลำดับ GAA ซ้ำทำให้เกิดการปิดการถอดรหัส (transcriptional silencing) ส่งผลให้ระดับ mRNA ของ FXN ลดลง 1) เมื่อขาดฟราทาซินจะเกิดสิ่งต่อไปนี้
การสะสมธาตุเหล็กในไมโตคอนเดรีย : เกิดจากการทำงานที่บกพร่องในการจัดการธาตุเหล็กการผลิต ATP ลดลง : เนื่องจากความผิดปกติของระบบหายใจโซ่คอมเพล็กซ์2) ROS เพิ่มขึ้น : ภาวะเครียดออกซิเดชัน สูงขึ้น1) 2)
ปมประสาทรากหลัง : มีขนาดเล็กและฝ่อ รากหลังมีลักษณะบางและสีเทาไขสันหลัง : เส้นผ่านศูนย์กลางลดลงทั่วทั้งส่วน โดยเฉพาะบริเวณทรวงอก เกิดการเสื่อมแบบก้าวหน้าของคอร์ติโคสไปนัลทรัคต์ สไปโนซีรีเบลลาร์ทรัคต์ และดอร์ซอลคอลัมน์
การขาดฟราทาซินทำให้ระดับธาตุเหล็กภายในเซลล์เปลี่ยนแปลง ทำให้เซลล์ปมประสาทจอประสาทตา (RGC ) อ่อนแอต่อภาวะเครียดออกซิเดชัน ผู้ป่วย FA สูงสุด 30% มีอาการทางจักษุวิทยา ทั้งวงจรก้านสมอง-ซีรีเบลลัมและเส้นประสาทตา ต่างได้รับความเสียหาย การลดลงของความหนาชั้นเส้นใยประสาทจอประสาทตา (RNFL ) สัมพันธ์โดยตรงกับการลดลงของการมองเห็น และความไวต่อคอนทราสต์ ข้อบกพร่องลานสายตาส่วนต้นที่ดำเนินจากรอบนอกเข้าสู่ศูนย์กลางสัมพันธ์กับการสูญเสีย RNFL ที่บันทึกด้วย OCT
โรคกล้ามเนื้อหัวใจหนาตัวผิดปกติ (hypertrophic cardiomyopathy) ทำให้น้ำหนักหัวใจเพิ่มขึ้น เนื้อเยื่อกล้ามเนื้อหัวใจอาจมีลักษณะภายนอกคล้ายหินอ่อน (marble-like)
ความผิดปกติของไมโทคอนเดรียเป็นสาเหตุพื้นฐานของโรคเบาหวานที่เกี่ยวข้องกับ FRDA 2)
ความผิดปกติของเซลล์เบตา : การผลิต ATP ที่บกพร่องทำให้การหลั่งอินซูลินลดลง และในที่สุดนำไปสู่การสูญเสียเซลล์เบตาความผิดปกติของการควบคุมการหลั่งของเซลล์อัลฟา : ภาวะกลูคากอนในเลือดสูงอย่างขัดแย้งเมื่อน้ำตาลในเลือดสูง และการตอบสนองของกลูคากอนไม่เพียงพอเมื่อน้ำตาลในเลือดต่ำ 2) กลไกการดื้ออินซูลิน : การนำกลูโคสเข้าสู่เซลล์ลดลงเนื่องจากความบกพร่องของ oxidative phosphorylation ในกล้ามเนื้อโครงร่าง การสะสมไขมันนอกตำแหน่งในตับและกล้ามเนื้อเนื่องจากความผิดปกติของเมแทบอลิซึมของไขมัน การรบกวนสัญญาณอินซูลินจากการอักเสบเรื้อรังและความเครียดออกซิเดชัน ความผิดปกติของการผลิตกลูโคสในตับจากระบบประสาทอัตโนมัติที่ผิดปกติ และการเสื่อมสภาพของเมแทบอลิซึมทุติยภูมิจากการไม่เคลื่อนไหวร่างกายและการฝ่อของกล้ามเนื้อเนื่องจากภาวะเสียการทรงตัว 2)
ผู้ที่มี Intragenic deletion มักมีแนวโน้มที่จะเริ่มมีอาการเร็วขึ้น ดำเนินโรคเร็วขึ้น และมีกล้ามเนื้อหัวใจผิดปกติรุนแรงกว่าเมื่อเทียบกับ biallelic expansion 1) หากการลบทำให้ start codon หายไป การผลิตโปรตีนจะหายไปโดยสิ้นเชิง 1)
Q
ทำไมโรคฟรีดไรค์อะแท็กเซียจึงทำให้เกิดอาการทางตา?
A
ความผิดปกติของการเผาผลาญธาตุเหล็กและความเครียดออกซิเดชันจากการขาดฟราทาซินทำให้เซลล์ปมประสาทจอประสาทตา เสียหาย การลดลงของความหนา RNFL สัมพันธ์โดยตรงกับการมองเห็น และความไวต่อคอนทราสต์ที่ลดลง ทำให้เกิดการสูญเสียลานสายตาแบบศูนย์กลางจากบริเวณรอบนอก นอกจากนี้ ความเสียหายต่อวงจรก้านสมองและสมองน้อยยังทำให้เกิดความผิดปกติของการเคลื่อนไหวของลูกตา เช่น อาตา การเคลื่อนไหวตามวัตถุผิดปกติ และการวัดการกระตุกของลูกตาผิดปกติ
เนื้อหาต่อไปนี้เป็นข้อมูลที่อยู่ในระยะวิจัยหรือการทดลองทางคลินิกเท่านั้น ไม่ใช่การรักษามาตรฐานที่สามารถรับได้ในโรงพยาบาลทั่วไป เป็นข้อมูลอ้างอิงสำหรับผู้เชี่ยวชาญเกี่ยวกับการพัฒนาทางการแพทย์ในอนาคต
การทดลองทางคลินิกของ Cooper et al. (2008) รายงานว่าการรักษาร่วมด้วย CoQ10 และวิตามินอีช่วยให้คะแนน ICARS ดีขึ้นอย่างมีนัยสำคัญในช่วงระยะเวลา 2 ปี2)
การทดลองแบบสุ่มและมีกลุ่มควบคุมด้วยยาหลอกแบบไขว้ของ Schöls et al. (2005) แสดงให้เห็นว่าการให้ L-carnitine ช่วยเพิ่มการผลิต ATP ในไมโตคอนเดรียอย่างมีนัยสำคัญ2)
Sureshkumar et al. (2025) รายงานกรณีผู้หญิงอายุ 32 ปีที่มีโรคเบาหวานที่เกี่ยวข้องกับ FA ซึ่งได้รับการรักษาด้วยการรวมกันของอินซูลิน, sitagliptin, L-carnitine, CoQ10, วิตามินอี, วิตามินบำรุงระบบประสาท, และ imeglimin2) ค่า HbA1c ลดลงจาก 13.3% เป็น 8.4% หลังจาก 17 เดือน และเป็น 6.9% หลังจาก 19 เดือน คะแนน ICARS ลดลงจาก 85 เป็น 71 (ลดลง 14 คะแนน) (HOMA2-IR: 4.5→1.2, HOMA2-%B: 5→60, MAGE: 120→70 mg/dL, CV: 43%→34.9%) ในขณะที่ประวัติธรรมชาติของโรคตาม SARA คาดว่าจะแย่ลงประมาณ 2.31 คะแนนใน 3 ปี แต่พบว่ามีความเบี่ยงเบนเชิงบวกประมาณ 16 คะแนน ถือเป็นรายงานผู้ป่วยรายแรกที่แสดงให้เห็นถึงการควบคุมระดับน้ำตาลในเลือดที่คงที่ในระยะยาวและการปรับปรุงภาวะเสียการทรงตัวร่วมกัน
เชื่อกันว่า imeglimin ออกฤทธิ์ผ่านการปรับปรุงการทำงานของห่วงโซ่การหายใจ ลดความเครียดออกซิเดชัน และส่งเสริมการสังเคราะห์ ATP/NAD+2)
การบำบัดด้วยเวกเตอร์ AAV : การทดลองทางคลินิกของ AAVrh.10hFXN ซึ่งมีเป้าหมายเพื่อนำยีน FXN ปกติเข้าสู่หัวใจและระบบประสาทกำลังดำเนินอยู่ และแสดงให้เห็นถึงการปรับปรุงในช่วงต้นของการแสดงออกของ FXN และเครื่องหมายของโรค (Munoz-Zuluaga et al. 2023)2) CRISPR-Cas9 : มีวัตถุประสงค์เพื่อกำจัดลำดับซ้ำ GAA โดยกำลังศึกษาอยู่ในเซลล์และแบบจำลองหนูที่ได้จาก YG8R (Ouellet et al. 2017)2) ความท้าทาย : จำเป็นต้องควบคุมการตอบสนองทางภูมิคุ้มกันและสร้างวิธีการส่งยีนที่ปลอดภัย2)
Aguilera และคณะ (2023) ระบุการขาดหายของยีนภายใน (intragenic deletion) ใหม่ที่ครอบคลุม 5’UTR และเอ็กซอน 1-2 ของยีน FXN โดยการตรวจตัวอย่างพ่อแม่ของผู้ป่วยที่เคยสันนิษฐานว่ามีการขยายตัวแบบ biallelic 1) จนถึงปัจจุบันมีการรายงานการขาดหายของยีนภายในเพียง 10 รายใน文献 แต่มีความเป็นไปได้ว่าอาจพบได้บ่อยกว่าที่คาด การวิเคราะห์ยีนเสริมด้วยวิธี MLPA และการตรวจตัวอย่างพ่อแม่ให้เป็นมาตรฐานถือเป็นความท้าทายในอนาคต
รายงานการตรวจพบครั้งแรกในแอฟริกาตะวันตกชี้ให้เห็นถึงความจำเป็นในการศึกษาแบบกลุ่มตัวอย่างขนาดใหญ่ในภูมิหลังทางชาติพันธุ์และภูมิศาสตร์ที่หลากหลาย3) การระบุตัวแปรที่ปรับเปลี่ยนโรคอาจเป็นเป้าหมายการรักษาในอนาคต
Aguilera C, Esteve-Garcia A, Casasnovas C, et al. Novel intragenic deletion within the FXN gene in a patient with typical phenotype of Friedreich ataxia: may be more prevalent than we think? BMC Med Genomics. 2023;16:312.
Sureshkumar P, Kumar SS, Cheriyan J, Masood A. Friedreich Ataxia and Related Diabetes: Therapeutic Approach Targeting Mitochondrial Dysfunction. JCEM Case Rep. 2025;3:luaf215.
Cissé CAK, Cissé L, Ba HO, et al. Friedreich ataxia in a family from Mali, West Africa. Clin Case Rep. 2021;9:e04065.