A ataxia de Friedreich (FRDA) é uma doença hereditária progressiva que afeta o sistema nervoso central e periférico. É considerada a ataxia hereditária autossômica recessiva mais frequente na população branca.
Em 1863, Nikolaus Friedreich descreveu casos de ataxia de início juvenil, escoliose e degeneração cardíaca familiar. Posteriormente, Pierre Marie distinguiu a FRDA de outras ataxias, estabelecendo o conceito da doença.
Epidemiologicamente, há grande variação regional, com prevalência relatada de 1:20.000 a 1:750.000. Em europeus, é de aproximadamente 1:21.0001); globalmente, estima-se entre 1:40.0002) e 1:50.0003). A frequência de portadores é de cerca de 1/701). A doença é mais frequente no sul da França, norte da Espanha e Irlanda, e menos comum na Escandinávia e Rússia. Na África subsaariana e no Hemisfério Oriental, é ainda mais rara, mas há casos confirmados geneticamente na África Ocidental (em famílias tuaregues com casamentos consanguíneos no Mali)3).
O padrão de herança é autossômico recessivo, com incidência igual entre homens e mulheres. A idade típica de início é em média 15,5 anos, geralmente antes dos 25 anos. A maioria dos casos ocorre entre 8 e 15 anos2). A expectativa de vida média é de 39 anos, sendo a principal causa de morte a cardiomiopatia2).
QQual a frequência da ataxia de Friedreich?
A
É a ataxia hereditária mais frequente na população branca, com prevalência variando de 1:20.000 a 1:750.000 conforme a região. Na Europa, estima-se que afete cerca de 1 em cada 21.000 pessoas1), e mundialmente, 1 em cada 40.000 a 50.0002)3). A frequência de portadores é de aproximadamente 1 em 701).
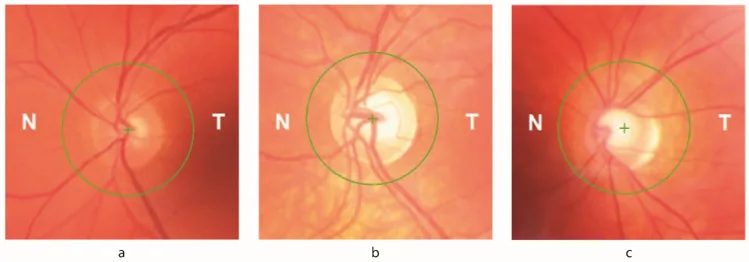
Petya Bogdanova-Mihaylova, Helena Maria Plapp, Hongying Chen et al. Longitudinal Assessment Using Optical Coherence Tomography in Patients with Friedreich’s Ataxia. Tomography. 2021 Dec 8; 7(4):915. Figure 1. PMCID: PMC8706975. License: CC BY.
Distúrbio da marcha: sintoma inicial mais comum. Começa com marcha instável em todas as direções.
Desajeitamento e tremor dos membros superiores: alguns casos iniciam-se com tremor nas mãos3).
Disartria: frequentemente surge dentro de 10 a 15 anos após o início da doença.
Sintomas sensoriais: Perda da sensibilidade profunda (propriocepção).
Sintomas visuais: Alguns pacientes apresentam diminuição da acuidade visual e da sensibilidade ao contraste, mas a maioria não apresenta sintomas visuais subjetivos no início.
A progressão dos sintomas motores varia entre os indivíduos, mas, em média, cerca de 8 anos após o início dos sintomas, o paciente perde a capacidade de andar sozinho, e em 11 a 15 anos, necessita de cadeira de rodas. Em casos com maior número de repetições GAA, há relatos de necessidade de cadeira de rodas em 5 anos 3).
Achados clínicos (achados confirmados pelo médico durante o exame)
Marcha atáxica: Marcha instável em todas as direções. Acompanhada de dismetria, hipotonia, disdiadococinesia e comprometimento do tempo de movimento coordenado 1).
Ausência de reflexos tendinosos profundos: A ausência dos reflexos patelar e aquileu é um item obrigatório nos critérios diagnósticos de Harding. A ausência de reflexos nos membros superiores também é frequente. Em alguns casos, podem ocorrer hiper-reflexia, espasticidade e marcha em tesoura 3).
Sinal de Babinski: Aparece como resposta plantar extensora.
Deformidades esqueléticas: escoliose (presente desde o início), pé cavo (pes cavus), lordose lombar3).
Nistagmo: devido a disfunção dos circuitos cerebelares e do tronco encefálico. Inclui nistagmo dependente da posição do olhar.
Movimentos sacádicos em onda quadrada (square-wave jerks): movimentos oculares involuntários e impulsivos que ocorrem durante a fixação.
Alteração do movimento de perseguição: o rastreamento visual torna-se fragmentado (perseguição sacádica).
Dismetria sacádica: ocorrência de sacadas hipométricas ou hipermétricas.
Atrofia óptica: observada ao exame de fundo de olho. Sinais oftalmológicos estão presentes em até 30% dos pacientes.
Redução da espessura da RNFL: detectada por OCT. Correlaciona-se diretamente com a diminuição da acuidade visual e da sensibilidade ao contraste.
Defeito de campo visual: defeito inicial que progride concentricamente a partir da periferia. Associado à perda de RNFL registrada por OCT.
QOs sintomas oculares aparecem sempre?
A
Na FRDA, até 30% dos pacientes apresentam sinais oftalmológicos, mas muitos não têm sintomas visuais subjetivos no início. Atrofia do nervo óptico, redução da espessura da RNFL e distúrbios da motilidade ocular são detectados por exames objetivos e correlacionam-se diretamente com a diminuição da acuidade visual e da sensibilidade ao contraste. Recomenda-se exame oftalmológico regular.
A causa da FRDA é a expansão da repetição de três bases GAA no íntron 1 do gene FXN (frataxina) no cromossomo 9. 96% dos casos são homozigotos para a expansão patogênica bialélica1). Os casos restantes são heterozigotos compostos com expansão GAA e mutação pontual, ou expansão GAA e deleção intrônica/de gene inteiro1).
A relação entre o número de repetições GAA e a doença é mostrada abaixo.
Classificação
Número de repetições GAA
Normal
5 a 33 repetições
Intermediário (equivalente a portador)
34 a 65 repetições
Patológico
66 repetições ou mais (em outros relatos, 90 ou mais)1)3)
Quanto maior o número de repetições GAA, mais precoce é o início e mais grave a doença. Em um caso com alelos extremamente longos de 999/766 repetições, o início ocorreu aos 11 anos e o paciente necessitou de cadeira de rodas em menos de 5 anos (mais rápido que os 10 anos típicos)3). Interrupções na sequência de repetições podem atrasar o início da doença1).
Heterozigotos compostos com deleção intrônica + expansão GAA tendem a ter início mais precoce, progressão mais rápida e cardiomiopatia mais grave do que aqueles com expansão bialélica, embora casos com curso típico também tenham sido relatados1).
Casamentos consanguíneos aumentam o risco de desenvolver a doença devido à sua natureza autossômica recessiva2)3). A frequência é maior em populações de ascendência europeia.
QSe o teste genético indicar homozigose, é possível que haja heterozigose composta?
A
A análise de fragmentos de PCR e TP-PCR não conseguem detectar deleções intrragênicas, portanto, uma aparente expansão bialélica pode ser, na verdade, uma expansão GAA + deleção intrragênica 1). Testes adicionais com MLPA (Amplificação Multiplex Dependente de Sondas de Ligação) e análise de amostras parentais são essenciais para um aconselhamento genético preciso.
Para o diagnóstico clínico, são utilizados os critérios de Harding.
Categoria
Principais itens
Itens obrigatórios
Início antes dos 25 anos, ataxia progressiva da marcha e dos membros, ausência de reflexos patelares e aquileus, neuropatia axonal, disartria (após 5 anos do início)
Achados adicionais (≥66%)
Escoliose, fraqueza muscular piramidal dos membros inferiores, arreflexia dos membros superiores, perda sensorial em fibras nervosas grossas, eletrocardiograma anormal
PCR + TP-PCR: Amplifica a repetição GAA no íntron 1 e detecta alelos expandidos.
MLPA (Amplificação de Sondas Dependente de Ligação Multiplex): Necessária para diagnosticar deleções/duplicações não detectáveis por análise de fragmentos ou TP-PCR1). Adicionada para excluir a possibilidade de uma aparente expansão bialélica ser na verdade um heterozigoto composto.
Análise por Southern blot: Combinada com PCR, permite diagnóstico com precisão >99%3).
Mostra neuropatia sensorial caracterizada pela ausência de potencial de ação nervoso sensorial (SNAP). O padrão de polineuropatia sensorial axonal é característico3).
Eletro-oculografia (EOG): usada para análise qualitativa e quantitativa dos movimentos oculares. Avalia sacadas guiadas visualmente (latência e amplitude), perseguição (ganho para diferentes velocidades do alvo), VOR (ganho para diferentes velocidades angulares da cabeça) e fixação (análise da forma de onda do nistagmo, latência de ondas quadradas).
Tomografia de coerência óptica (OCT): mede quantitativamente a espessura da camada de fibras nervosas da retina (CFNR). Pode confirmar correlação com redução da acuidade visual e sensibilidade ao contraste.
Campimetria: avalia padrão de defeito que progride concentricamente da periferia.
Exame de fundo de olho: verifica presença de atrofia óptica.
Eletrocardiograma e ecocardiograma: avaliam cardiomiopatia, espessura da parede ventricular esquerda, fração de ejeção e função diastólica2). É necessário monitoramento regular.
Exames relacionados à glicemia2): glicemia de jejum, HbA1c, peptídeo C, insulina de jejum, autoanticorpos das ilhotas pancreáticas (GAD65, IA-2, ZnT8), HOMA2-IR e HOMA2-%B para detecção precoce e classificação do diabetes.
Escalas de avaliação clínica: FARS (Friedreich Ataxia Rating Scale), ICARS e SARA para quantificar sintomas neurológicos2). Na SARA, foi relatada uma piora anual de 0,77 pontos (EP 0,06) na evolução natural2).
Ressonância magnética do cérebro e medula espinhal: avaliação de atrofia cerebelar (houve caso com aparecimento 3 anos após o início3)) e sinais degenerativos da medula espinhal.
Atualmente, não existe tratamento curativo, e o tratamento é centrado no manejo sintomático e das complicações por meio de uma abordagem multidisciplinar.
Manejo da escoliose: para casos leves a moderados, uso de órtese; para casos graves, considerar cirurgia.
Gerenciamento do pé cavo: Injeção de toxina botulínica no gastrocnêmio e alongamento do tendão de Aquiles para melhorar a mobilidade.
Reabilitação: Fisioterapia, terapia ocupacional e fonoaudiologia para manutenção da função.
Oftalmologia
Cuidados para baixa visão: Para pacientes com sintomas visuais devido à atrofia do nervo óptico e atrofia retiniana, fornecer lupas, ajuste de iluminação e orientação para atividades diárias.
Tratamento sintomático dos distúrbios dos movimentos oculares: Para nistagmo dependente da posição ocular, usar óculos prismáticos (adicionar potência prismática igual em ambos os olhos na direção da posição ocular que piora). Para nistagmo vertical, nistagmo periódico alternante e movimentos oculares sacádicos, administrar agonistas GABA_B.
Exemplo de prescrição (agonista GABA_B): Comprimidos de Gabarone (5 mg) 3 a 6 comprimidos, 1 a 3 vezes ao dia.
O diabetes associado à FRDA tem disfunção mitocondrial subjacente, portanto, é necessário cuidado na seleção de medicamentos2).
Medicamentos a evitar: Metformina e tiazolidinedionas devem ser evitados porque inibem o complexo I mitocondrial2). Sulfonilureias apresentam risco de estresse das células beta e hipoglicemia.
Medicamentos hipoglicemiantes recomendados: Inibidores da DPP-4 (ex.: sitagliptina 100 mg/dia) e análogos do GLP-1 são preferíveis2). Imeglimina (500 mg 2×/dia) pode ser útil como novo hipoglicemiante direcionado à função mitocondrial2).
Terapia com insulina: Iniciar com aproximadamente 0,5 U/kg/dia2).
Terapia adjuvante direcionada à mitocôndria: Relata-se o uso combinado de L-carnitina 500 mg/dia, CoQ10 100 mg/dia e vitamina E 400 UI/dia2).
Em um relato de caso com o regime acima, a HbA1c melhorou de 13,3% para 8,4% (após 17 meses) e depois para 6,9% (após mais 19 meses), e o ICARS melhorou 14 pontos, de 85 para 712).
QQual tratamento é adequado para o diabetes associado à ataxia de Friedreich?
A
Devido à disfunção mitocondrial subjacente, metformina e tiazolidinedionas devem ser evitados 2). Inibidores da DPP-4 (como sitagliptina) e análogos do GLP-1 são recomendados. Terapias adjuvantes direcionadas às mitocôndrias, como L-carnitina, CoQ10 e vitamina E, também podem ser úteis 2).
6. Fisiopatologia e mecanismos detalhados da doença
A frataxina é uma proteína localizada na membrana mitocondrial interna. Está envolvida no metabolismo do ferro (armazenamento de ferro e montagem de clusters de ferro-enxofre) e é essencial para o funcionamento normal da cadeia respiratória mitocondrial 1).
A expansão da repetição GAA causa silenciamento transcricional, reduzindo os níveis de mRNA da FXN 1). A deficiência de frataxina leva ao seguinte:
Acúmulo de ferro mitocondrial: devido à função prejudicada de processamento de ferro.
Redução da produção de ATP: devido à disfunção dos complexos da cadeia respiratória2).
Gânglios da raiz dorsal: pequenos e atrofiados, com raízes dorsais finas e acinzentadas.
Medula espinhal: diâmetro reduzido em toda a extensão, especialmente na região torácica. Ocorre degeneração progressiva do trato corticospinal, trato espinocerebelar e coluna posterior.
A deficiência de frataxina altera os níveis de ferro intracelular, tornando as células ganglionares da retina (CGR) vulneráveis ao estresse oxidativo. Até 30% dos pacientes com AF apresentam sinais oftalmológicos. Tanto os circuitos do tronco encefálico e cerebelo quanto o nervo óptico são afetados. A redução da espessura da CFNR correlaciona-se diretamente com a diminuição da acuidade visual e da sensibilidade ao contraste, e os defeitos iniciais do campo visual, que progridem concentricamente a partir da periferia, estão associados à perda da CFNR documentada pela OCT.
A disfunção mitocondrial é a causa subjacente do diabetes associado à FRDA2).
Disfunção das células beta: A produção prejudicada de ATP reduz a secreção de insulina, levando eventualmente à perda de células beta.
Desregulação da secreção de células alfa: Hiperglucagonemia paradoxal durante hiperglicemia e resposta inadequada do glucagon durante hipoglicemia2).
Mecanismos de resistência à insulina: Captação reduzida de glicose devido à fosforilação oxidativa prejudicada no músculo esquelético, acúmulo de gordura ectópica no fígado e músculo devido ao metabolismo lipídico alterado, sinalização de insulina prejudicada por inflamação crônica e estresse oxidativo, produção hepática anormal de glicose devido à neuropatia autonômica e deterioração metabólica secundária devido à inatividade física e atrofia muscular causadas por ataxia2).
Portadores de deleção intra-gênica tendem a apresentar início mais precoce, progressão rápida e cardiomiopatia mais grave do que aqueles com expansão bialélica1). Se a deleção remover o códon de início, a produção de proteína é completamente perdida1).
QPor que a ataxia de Friedreich causa sintomas oculares?
A
A deficiência de frataxina leva a anormalidades no metabolismo do ferro e estresse oxidativo, que danificam as células ganglionares da retina. A redução da espessura da RNFL correlaciona-se diretamente com a diminuição da acuidade visual e da sensibilidade ao contraste, resultando em defeitos de campo visual que progridem concentricamente da periferia. Além disso, danos nos circuitos do tronco cerebral e cerebelo causam anormalidades nos movimentos oculares, como nistagmo, distúrbios de movimentos oculares de perseguição e dismetria sacádica.
7. Pesquisas recentes e perspectivas futuras (relatos em fase de pesquisa)
O ensaio clínico de Cooper et al. (2008) relatou que a terapia combinada de CoQ10 e vitamina E melhorou significativamente o escore ICARS ao longo de dois anos2).
O estudo cruzado randomizado controlado por placebo de Schöls et al. (2005) mostrou que a administração de L-carnitina melhorou significativamente a produção mitocondrial de ATP2).
Sureshkumar et al. (2025) administraram uma combinação de insulinoterapia, sitagliptina, L-carnitina, CoQ10, vitamina E, vitaminas neurotrópicas e imeglimina a uma mulher de 32 anos com diabetes relacionada à FA2). A HbA1c melhorou de 13,3% para 8,4% após 17 meses e para 6,9% após 19 meses, e o ICARS melhorou 14 pontos, de 85 para 71 (HOMA2-IR: 4,5→1,2; HOMA2-%B: 5→60; MAGE: 120→70 mg/dL; CV: 43%→34,9%). Considerando que a história natural prevê uma piora de aproximadamente 2,31 pontos em 3 anos pela SARA, foi observado um desvio positivo de cerca de 16 pontos. Este é considerado o primeiro caso a relatar a obtenção simultânea de estabilização glicêmica de longo prazo e melhora da ataxia.
Acredita-se que a imeglimina atue melhorando a atividade da cadeia respiratória, reduzindo o estresse oxidativo e promovendo a síntese de ATP/NAD+2).
Terapia com vetor AAV: O ensaio clínico do AAVrh.10hFXN, que visa a introdução do gene FXN normal no coração e no sistema nervoso, está em andamento, com melhora precoce da expressão de FXN e de marcadores da doença demonstrada (Munoz-Zuluaga et al. 2023)2).
CRISPR-Cas9: visa a remoção da sequência repetitiva GAA, e está sendo estudada em células e modelos de camundongos YG8R (Ouellet et al. 2017)2).
Desafio: É necessário controlar a resposta imune e estabelecer um método seguro de entrega de genes2).
Aguilera et al. (2023) identificaram uma nova deleção intra-gênica envolvendo a 5’UTR e os éxons 1-2 do gene FXN por meio de testes em amostras parentais de um paciente que se presumia ter expansão bialélica 1). Até o momento, apenas 10 deleções intra-gênicas foram relatadas na literatura, mas sugere-se que sua frequência possa ser maior. A padronização de análises genéticas complementares, como MLPA, e de testes em amostras parentais são desafios futuros.
O relato do primeiro caso confirmado na África Ocidental indica a necessidade de estudos de coorte em larga escala com diversas origens étnicas e geográficas3). A identificação de variantes modificadoras da doença pode se tornar alvos terapêuticos futuros.
Aguilera C, Esteve-Garcia A, Casasnovas C, et al. Novel intragenic deletion within the FXN gene in a patient with typical phenotype of Friedreich ataxia: may be more prevalent than we think? BMC Med Genomics. 2023;16:312.
Sureshkumar P, Kumar SS, Cheriyan J, Masood A. Friedreich Ataxia and Related Diabetes: Therapeutic Approach Targeting Mitochondrial Dysfunction. JCEM Case Rep. 2025;3:luaf215.
Cissé CAK, Cissé L, Ba HO, et al. Friedreich ataxia in a family from Mali, West Africa. Clin Case Rep. 2021;9:e04065.
Copie o texto do artigo e cole no assistente de IA de sua preferência.
Artigo copiado para a área de transferência
Abra um assistente de IA abaixo e cole o texto copiado na conversa.