La ataxia de Friedreich (FRDA) es una enfermedad hereditaria progresiva que afecta al sistema nervioso central y periférico. Es la ataxia hereditaria autosómica recesiva más frecuente en la población blanca.
En 1863, Nikolaus Friedreich describió casos de ataxia de inicio juvenil, escoliosis y degeneración cardíaca familiar. Posteriormente, Pierre Marie diferenció la FRDA de otras ataxias, estableciendo el concepto de la enfermedad.
Epidemiológicamente, existe una gran variabilidad regional, con una prevalencia reportada de 1:20.000 a 1:750.000. En europeos, se estima en aproximadamente 1:21.0001), y a nivel mundial, entre 1:40.0002) y 1:50.0003). La frecuencia de portadores se estima en aproximadamente 1/701). Es más frecuente en el sur de Francia, norte de España e Irlanda, y menos en Escandinavia y Rusia. En el África subsahariana y el hemisferio oriental es aún menos frecuente, aunque se han reportado casos confirmados genéticamente en África occidental (en familias tuareg con matrimonios consanguíneos en Malí)3).
El patrón de herencia es autosómico recesivo, y la incidencia es igual en hombres y mujeres. La edad típica de inicio es alrededor de los 15,5 años, y la mayoría de los casos se presentan antes de los 25 años. La edad de inicio más frecuente es entre los 8 y 15 años2). La esperanza de vida media es de 39 años, y la principal causa de muerte es la miocardiopatía2).
Q¿Con qué frecuencia ocurre la ataxia de Friedreich?
A
Es la ataxia hereditaria más frecuente en la población blanca, con una prevalencia que varía según la región, entre 1:20.000 y 1:750.000. En Europa, afecta aproximadamente a 1 de cada 21.000 personas1), y a nivel mundial se estima en 1 de cada 40.000 a 50.0002)3). La frecuencia de portadores se estima en aproximadamente 1 de cada 701).
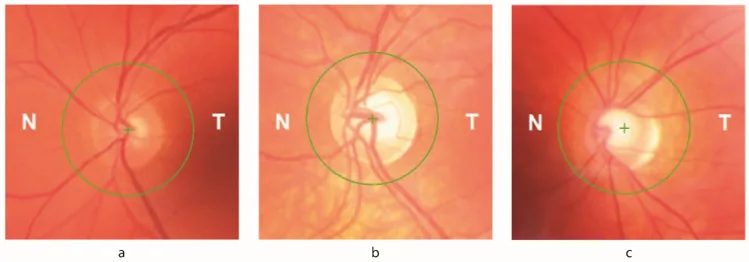
Petya Bogdanova-Mihaylova, Helena Maria Plapp, Hongying Chen et al. Longitudinal Assessment Using Optical Coherence Tomography in Patients with Friedreich’s Ataxia. Tomography. 2021 Dec 8; 7(4):915. Figure 1. PMCID: PMC8706975. License: CC BY.
Alteración de la marcha: síntoma inicial más frecuente. Comienza con una marcha inestable en todas las direcciones.
Torpeza y temblor en miembros superiores: en algunos casos, el temblor de manos puede ser el síntoma inicial3).
Disartria: suele aparecer dentro de los 10 a 15 años posteriores al inicio.
Síntomas sensoriales: se produce pérdida de la sensibilidad profunda (propiocepción).
Síntomas visuales: algunos pacientes presentan disminución de la agudeza visual y de la sensibilidad al contraste, pero la mayoría no presenta síntomas visuales subjetivos en las etapas iniciales.
La progresión de los síntomas motores varía entre individuos, pero en promedio, aproximadamente 8 años después del inicio de los síntomas, el paciente no puede caminar sin ayuda, y entre 11 y 15 años requiere silla de ruedas. Se ha reportado que en casos con un número elevado de repeticiones GAA, la necesidad de silla de ruedas puede ocurrir en 5 años 3).
Hallazgos clínicos (hallazgos confirmados por el médico durante el examen)
Marcha atáxica: marcha inestable en todas las direcciones. Se acompaña de dismetría, hipotonía, disdiadococinesia y alteración del tiempo de movimientos coordinados 1).
Ausencia de reflejos tendinosos profundos: la ausencia del reflejo rotuliano y del tendón de Aquiles es un criterio obligatorio en los criterios diagnósticos de Harding. La ausencia de reflejos en las extremidades superiores también es frecuente. En algunos casos se observa hiperreflexia, espasticidad y marcha en tijera 3).
Signo de Babinski: aparece como una respuesta plantar extensora.
Deformidad esquelética: escoliosis (presente desde el inicio), pie cavo (pes cavus), lordosis lumbar 3).
Nistagmo: debido a disfunción del circuito cerebeloso-troncoencefálico. Incluye nistagmo dependiente de la posición ocular.
Sacudidas de onda cuadrada (square-wave jerks): movimientos oculares sacádicos involuntarios que interfieren durante la fijación.
Alteración de los movimientos de persecución: el seguimiento del objetivo se vuelve escalonado (persecución anormal).
Dismetría sacádica (saccadic dysmetria): aparición de sacadas hipermétricas o hipométricas.
Atrofia óptica: observable en el examen de fondo de ojo. Hasta un 30% de los pacientes presentan signos oftalmológicos.
Reducción del grosor de la capa de fibras nerviosas de la retina (RNFL) : detectada mediante OCT. Se correlaciona directamente con la disminución de la agudeza visual y la sensibilidad al contraste.
Defecto del campo visual : defecto visual temprano que progresa concéntricamente desde la periferia. Se asocia con la pérdida de RNFL registrada por OCT.
Q¿Los síntomas oculares siempre aparecen?
A
En la FRDA, hasta un 30% de los pacientes presentan signos oculares, pero muchos pacientes carecen de síntomas visuales subjetivos en las etapas iniciales. La atrofia del nervio óptico, la reducción del grosor de la RNFL y los trastornos de la motilidad ocular se detectan mediante exámenes objetivos y se correlacionan directamente con la disminución de la agudeza visual y la sensibilidad al contraste. Se recomiendan exámenes oftalmológicos periódicos.
La causa de la FRDA es la expansión de la repetición de tres bases GAA en el intrón 1 del gen FXN (frataxina) en el cromosoma 9. El 96% de los casos son homocigotos para la expansión patogénica bialélica1). Los casos restantes son heterocigotos compuestos con una expansión GAA y una mutación puntual, o una expansión GAA y una deleción intrónica o de todo el gen1).
La relación entre el número de repeticiones GAA y la enfermedad se muestra a continuación.
Clasificación
Número de repeticiones GAA
Normal
5–33 repeticiones
Intermedio (equivalente a portador)
34–65 repeticiones
Patológico
66 repeticiones o más (según otros informes, 90 o más) 1)3)
Cuanto mayor es el número de repeticiones GAA, más temprana es la edad de inicio y más grave tiende a ser la enfermedad. En un caso con repeticiones alélicas extremadamente largas de 999/766, la enfermedad comenzó a los 11 años y se requirió silla de ruedas en menos de 5 años (más rápido que los 10 años típicos) 3). Las interrupciones dentro de la secuencia de repeticiones pueden retrasar la edad de inicio 1).
Se considera que los heterocigotos compuestos con deleción intrónica + expansión GAA tienden a tener un inicio más temprano, progresión más rápida y miocardiopatía más grave que las expansiones bialélicas, aunque también se han reportado casos con un curso típico 1).
El matrimonio consanguíneo aumenta el riesgo de enfermedad debido a la naturaleza autosómica recesiva de la enfermedad 2)3). La frecuencia es mayor en poblaciones de ascendencia europea.
QSi la prueba genética indica homocigosis, ¿es posible que sea un heterocigoto compuesto?
A
El análisis de fragmentos de PCR y TP-PCR no pueden detectar deleciones intragénicas, por lo que una aparente expansión bialélica podría ser en realidad una expansión GAA más una deleción intragénica 1). Las pruebas adicionales con MLPA (amplificación de sondas dependiente de ligación múltiple) y el análisis de muestras parentales son esenciales para un asesoramiento genético preciso.
Para el diagnóstico clínico se utilizan los criterios de Harding.
Categoría
Ítems principales
Ítems obligatorios
Inicio antes de los 25 años, ataxia progresiva de la marcha y extremidades, ausencia de reflejos rotulianos y aquíleos, neuropatía axonal, disartria (después de 5 años del inicio)
Hallazgos adicionales (≥66%)
Escoliosis, debilidad piramidal de miembros inferiores, arreflexia de miembros superiores, pérdida sensorial de fibras nerviosas gruesas, ECG anormal
PCR + TP-PCR: Amplifica la repetición GAA en el intrón 1 y detecta alelos expandidos.
MLPA (Amplificación de Sondas Dependiente de Ligación Múltiple): Necesaria para diagnosticar deleciones/duplicaciones no detectables por análisis de fragmentos o TP-PCR1). Se añade para descartar que una aparente expansión bialélica sea en realidad un compuesto heterocigoto.
Análisis de Southern blot: Combinado con PCR permite un diagnóstico con precisión >99%3).
Muestra una neuropatía sensorial caracterizada por la ausencia del potencial de acción nervioso sensorial (SNAP). El patrón de polineuropatía sensorial axonal es característico3).
Electrooculografía (EOG): se utiliza para el análisis cualitativo y cuantitativo de los movimientos oculares. Evalúa sacadas guiadas visualmente (latencia y amplitud), persecución (ganancia para diferentes velocidades del objetivo), VOR (ganancia para diferentes velocidades angulares de la cabeza) y fijación (análisis de la forma de onda del nistagmo, latencia de las ondas cuadradas).
Tomografía de coherencia óptica (OCT): mide cuantitativamente el grosor de la capa de fibras nerviosas de la retina (CFNR). Puede confirmar la correlación con la disminución de la agudeza visual y la sensibilidad al contraste.
Campimetría: evalúa el patrón de defecto que progresa concéntricamente desde la periferia.
Fondo de ojo: confirma la presencia o ausencia de atrofia del nervio óptico.
Cuando el síntoma principal es la alteración de los movimientos oculares, es necesario realizar un diagnóstico diferencial con las siguientes enfermedades.
Casos esporádicos de degeneración espinocerebelosa (SCD): incluye la atrofia multisistémica como parte del cuadro.
Malformación de Chiari: caracterizada por oscilopsia debido a nistagmo descendente.
SCD hereditaria: como los tipos SCA 6, 31 y 3.
Trastornos adquiridos: lesiones cerebelosas o del tronco encefálico por inflamación, tumor o trastorno vascular.
Electrocardiograma y ecocardiograma: evalúan miocardiopatía, grosor de la pared ventricular izquierda, fracción de eyección y función diastólica2). Se requiere monitorización periódica.
Pruebas relacionadas con la glucosa2): glucosa en ayunas, HbA1c, péptido C, insulina en ayunas, autoanticuerpos de islotes pancreáticos (GAD65, IA-2, ZnT8), HOMA2-IR, HOMA2-%B para la detección temprana y clasificación del tipo de diabetes.
Escalas de evaluación clínica: FARS (Friedreich Ataxia Rating Scale), ICARS, SARA para la evaluación cuantitativa de los síntomas neurológicos2). Con SARA se ha reportado un empeoramiento anual de 0.77 puntos (EE 0.06) en el curso natural2).
RM cerebral y espinal: evaluación de la atrofia cerebelosa (con casos que aparecen 3 años después del inicio3)) y hallazgos degenerativos de la médula espinal.
Actualmente no existe un tratamiento curativo, y el manejo se centra en el tratamiento sintomático y el control de complicaciones mediante un enfoque multidisciplinario.
Manejo de la escoliosis: para casos leves a moderados, ortesis; para casos graves, considerar cirugía.
Manejo del pie cavo: inyección de toxina botulínica en el gastrocnemio y estiramiento del tendón de Aquiles para mejorar la movilidad.
Rehabilitación: fisioterapia, terapia ocupacional y logopedia para mantener la función.
Oftalmología
Cuidado de baja visión: uso de lupas, ajuste de iluminación y asesoramiento en actividades diarias para pacientes con síntomas visuales por atrofia del nervio óptico y retiniana.
Tratamiento sintomático de trastornos del movimiento ocular: para el nistagmo dependiente de la posición ocular, se utilizan gafas prismáticas (añadiendo la misma potencia prismática en ambos ojos en la dirección que empeora el nistagmo). Para el nistagmo vertical, el nistagmo periódico alternante y los movimientos oculares sacádicos, se administran agonistas GABA_B.
Ejemplo de prescripción (agonista GABA_B): 3-6 comprimidos de Gabarón (5 mg), divididos en 1-3 tomas.
La diabetes asociada a FRDA se debe a una disfunción mitocondrial subyacente, por lo que se requiere precaución en la selección de fármacos2).
Medicamentos que deben evitarse: la metformina y las tiazolidinedionas inhiben el complejo mitocondrial I, por lo que deben evitarse2). Las sulfonilureas conllevan riesgo de estrés de células beta e hipoglucemia.
Fármacos hipoglucemiantes recomendados: se prefieren los inhibidores de DPP-4 (p. ej., sitagliptina 100 mg/día) y los análogos de GLP-12). La imeglimina (500 mg × 2/día) puede ser útil como nuevo hipoglucemiante dirigido a la función mitocondrial2).
Terapia con insulina: iniciar con una dosis de 0,5 U/kg/día como referencia2).
Terapia coadyuvante dirigida a la mitocondria: se ha reportado el uso combinado de L-carnitina 500 mg/día, CoQ10 100 mg/día y vitamina E 400 UI/día2).
En un reporte de caso con el régimen anterior, la HbA1c mejoró de 13,3% a 8,4% (tras 17 meses) y luego a 6,9% (tras 19 meses adicionales), y la ICARS mejoró 14 puntos, de 85 a 712).
Q¿Qué tratamiento es adecuado para la diabetes asociada a la ataxia de Friedreich?
A
Debido a la disfunción mitocondrial subyacente, deben evitarse la metformina y las tiazolidinedionas 2). Se recomiendan los inhibidores de la DPP-4 (como sitagliptina) y los análogos de GLP-1. Las terapias coadyuvantes dirigidas a la mitocondria, como L-carnitina, CoQ10 y vitamina E, también pueden ser útiles 2).
6. Fisiopatología y mecanismo detallado de la enfermedad
La frataxina es una proteína localizada en la membrana mitocondrial interna. Participa en el metabolismo del hierro (almacenamiento de hierro y ensamblaje de los grupos hierro-azufre) y es esencial para el funcionamiento normal de la cadena respiratoria mitocondrial 1).
La expansión de la repetición GAA provoca silenciamiento transcripcional, reduciendo los niveles de ARNm de FXN 1). La deficiencia de frataxina produce lo siguiente:
Acumulación de hierro mitocondrial: debido a la función deficiente del procesamiento del hierro.
Disminución de la producción de ATP: por disfunción de los complejos de la cadena respiratoria2).
Ganglios de la raíz dorsal: pequeños y atróficos, las raíces dorsales son delgadas y de color gris.
Médula espinal: diámetro reducido en toda su extensión, especialmente en la región torácica. Degeneración progresiva del tracto corticoespinal, espinocerebeloso y cordón posterior.
La deficiencia de frataxina altera los niveles de hierro intracelular, haciendo que las células ganglionares de la retina (CGR) sean vulnerables al estrés oxidativo. Hasta un 30% de los pacientes con AF presentan signos oftalmológicos. Tanto el tronco encefálico y los circuitos cerebelosos como el nervio óptico se ven afectados. La reducción del grosor de la capa de fibras nerviosas de la retina (CFNR) se correlaciona directamente con la disminución de la agudeza visual y la sensibilidad al contraste. Los defectos iniciales del campo visual, que progresan concéntricamente desde la periferia, se asocian con la pérdida de CFNR registrada por OCT.
La miocardiopatía hipertrófica aumenta el peso del corazón. El tejido miocárdico puede tener una apariencia macroscópica «similar al mármol» (marble-like).
La disfunción mitocondrial es la causa subyacente de la diabetes asociada a FRDA2).
Disfunción de las células beta: la producción deficiente de ATP reduce la secreción de insulina y, finalmente, conduce a la pérdida de células beta.
Alteración en la secreción de células α: hiperglucagonemia paradójica durante la hiperglucemia y respuesta deficiente del glucagón durante la hipoglucemia2).
Mecanismo de resistencia a la insulina: disminución de la captación de glucosa debido a una alteración de la fosforilación oxidativa en el músculo esquelético, acumulación de grasa ectópica en el hígado y el músculo por trastornos del metabolismo lipídico, alteración de la señalización de la insulina por inflamación crónica y estrés oxidativo, producción anormal de glucosa hepática por disfunción autonómica, y empeoramiento metabólico secundario por inactividad física y atrofia muscular debido a ataxia2).
Los portadores de una deleción intrágénica tienden a presentar inicio más temprano, progresión rápida y miocardiopatía más grave en comparación con la expansión bialélica 1). Si la deleción elimina el codón de inicio, la producción de proteína se pierde por completo 1).
Q¿Por qué aparecen síntomas oculares en la ataxia de Friedreich?
A
La deficiencia de frataxina causa anomalías en el metabolismo del hierro y estrés oxidativo que dañan las células ganglionares de la retina. La reducción del grosor de la capa de fibras nerviosas de la retina (RNFL) se correlaciona directamente con la disminución de la agudeza visual y la sensibilidad al contraste, y produce defectos del campo visual que progresan de forma concéntrica desde la periferia. Además, la disfunción de los circuitos del tronco encefálico y el cerebelo provoca anomalías en los movimientos oculares, como nistagmo, alteraciones en el seguimiento ocular y dismetría sacádica.
7. Investigación reciente y perspectivas futuras (informes en fase de investigación)
El ensayo clínico de Cooper et al. (2008) informó que la terapia combinada de CoQ10 y vitamina E mejoró significativamente la puntuación ICARS durante dos años2).
El estudio cruzado aleatorizado controlado con placebo de Schöls et al. (2005) mostró que la administración de L-carnitina mejoró significativamente la producción mitocondrial de ATP2).
Sureshkumar et al. (2025) administraron una combinación de insulinoterapia, sitagliptina, L-carnitina, CoQ10, vitamina E, vitaminas neurotrópicas e imeglimina a una mujer de 32 años con diabetes asociada a FA2). La HbA1c mejoró del 13,3% al 8,4% después de 17 meses y al 6,9% después de 19 meses, y la ICARS mejoró 14 puntos, de 85 a 71 (HOMA2-IR: 4,5→1,2; HOMA2-%B: 5→60; MAGE: 120→70 mg/dL; CV: 43%→34,9%). Mientras que la historia natural según SARA predice un empeoramiento de aproximadamente 2,31 puntos en 3 años, se observó una desviación positiva de aproximadamente 16 puntos. Se considera el primer caso reportado de lograr simultáneamente estabilización glucémica a largo plazo y mejora de la ataxia.
Se cree que la imeglimina actúa mejorando la actividad de la cadena respiratoria, reduciendo el estrés oxidativo y promoviendo la síntesis de ATP/NAD+2).
Terapia con vectores AAV: AAVrh.10hFXN se encuentra en ensayos clínicos en curso para la introducción del gen FXN normal en el corazón y el sistema nervioso, mostrando una mejora temprana en la expresión de FXN y los marcadores de la enfermedad (Munoz-Zuluaga et al. 2023)2).
CRISPR-Cas9: Dirigido a eliminar las repeticiones GAA, se está investigando en células derivadas de YG8R y modelos de ratón (Ouellet et al. 2017)2).
Desafíos: Es necesario controlar la respuesta inmunitaria y establecer métodos seguros de administración génica2).
Aguilera et al. (2023) identificaron una nueva deleción intrónica que incluye la 5’UTR y los exones 1-2 del gen FXN mediante el análisis de muestras parentales de un paciente con presunta expansión bialélica1). Aunque solo se han reportado 10 casos de deleciones intrónicas en la literatura, se sugiere que podrían ser más frecuentes. La estandarización del análisis genético complementario con MLPA y el análisis de muestras parentales son desafíos futuros.
El primer caso confirmado en África Occidental indica la necesidad de estudios de cohortes a gran escala en diversos contextos étnicos y geográficos3). La identificación de variantes modificadoras de la enfermedad podría ser un objetivo terapéutico futuro.
Aguilera C, Esteve-Garcia A, Casasnovas C, et al. Novel intragenic deletion within the FXN gene in a patient with typical phenotype of Friedreich ataxia: may be more prevalent than we think? BMC Med Genomics. 2023;16:312.
Sureshkumar P, Kumar SS, Cheriyan J, Masood A. Friedreich Ataxia and Related Diabetes: Therapeutic Approach Targeting Mitochondrial Dysfunction. JCEM Case Rep. 2025;3:luaf215.
Cissé CAK, Cissé L, Ba HO, et al. Friedreich ataxia in a family from Mali, West Africa. Clin Case Rep. 2021;9:e04065.
Copia el texto del artículo y pégalo en el asistente de IA que prefieras.
Artículo copiado al portapapeles
Abre un asistente de IA abajo y pega el texto copiado en el chat.