弗里德赖希共济失调(FRDA)是白种人群中最常见的常染色体隐性遗传 性共济失调。
病因为第9号染色体上FXN基因内含子1中GAA三核苷酸重复序列的纯合性扩展,占病例的96%。
典型发病年龄为8至15岁,首发症状以步态障碍最为常见。
合并多器官损害,包括心肌病(高达63%)、糖尿病(5%至40%)及视神经萎缩 。
高达30%的患者出现眼科体征(视神经萎缩 、眼球震颤 、眼球运动障碍 )。
尚无根治性治疗,基础治疗为多学科协作的对症治疗和并发症管理。
平均寿命为39岁,主要死因为心肌病。
弗里德赖希共济失调(Friedreich Ataxia; FRDA)是一种进行性遗传性疾病,累及中枢和周围神经系统。在白人人群中,它是最常见的常染色体隐性遗传 性共济失调。
1863年,尼古拉斯·弗里德赖希报道了青少年发病的共济失调、脊柱侧弯和家族性心脏变性。后来,皮埃尔·玛丽将FRDA与其他共济失调区分开来,确立了该疾病的概念。
流行病学上,地区差异很大,患病率报告为1:20,000至1:750,000。在欧洲人中,约为1:21,0001) ;全球估计为1:40,0002) 至1:50,0003) 。携带者频率约为1/701) 。在南法国、北西班牙和爱尔兰频率较高,在斯堪的纳维亚和俄罗斯较低。在撒哈拉以南非洲和东半球则更低,但在西非(马里共和国图阿雷格族近亲结婚家系)也有遗传学确诊的报道3) 。
遗传方式为常染色体隐性,男女发病率相等。典型发病年龄平均为15.5岁,多数在25岁前发病,以8至15岁发病多见2) 。平均寿命为39岁,主要死因为心肌病2) 。
Q
弗里德赖希共济失调的发生频率是多少?
A
在白人群体中,弗里德赖希共济失调是最常见的遗传性共济失调,患病率因地区而异,范围在1:20,000至1:750,000之间。在欧洲,约每21,000人中有1人1) ,全球估计为每40,000至50,000人中有1人2) 3) 。携带者频率约为1/701) 。
Petya Bogdanova-Mihaylova, Helena Maria Plapp, Hongying Chen et al. Longitudinal Assessment Using Optical Coherence Tomography in Patients with Friedreich’s Ataxia. Tomography. 2021 Dec 8; 7(4):915. Figure 1. PMCID: PMC8706975. License: CC BY.
图:弗里德赖希共济失调患者常见的高弓足和锤状趾畸形
步态障碍 :最常见的首发症状。始于全方向性不稳定步态。上肢笨拙与震颤 :部分病例以手部震颤为首发症状3) 。构音障碍 :常在发病后10至15年内出现。感觉症状 :出现深部感觉(本体感觉)丧失。视觉症状 :部分患者出现视力 下降和对比敏感度 下降,但大多数患者在早期没有视觉自觉症状。
运动症状的进展存在个体差异,但从首发症状起平均约8年无法独立行走,11至15年需要轮椅。有报告称,GAA重复次数较多时,5年内就需要轮椅3) 。
全身神经所见
共济失调步态 :全方位的不稳定步态。伴有辨距障碍、肌张力低下、轮替运动障碍和协同运动时间障碍1) 。
深部腱反射消失 :膝腱反射和跟腱反射消失是Harding诊断标准的必需项目。上肢反射消失也常见。部分病例出现反射亢进、痉挛和剪刀步态3) 。
巴宾斯基征 :表现为伸性跖反射。
骨骼畸形 :脊柱侧弯(早期出现)、凹足(pes cavus)、腰椎后凸3) 。
全身并发症 :肥厚型心肌病(高达63%)、糖尿病(5-40%)2) 。
神经眼科表现
眼球震颤 (nystagmus)眼球震颤 。
方波跳动(square-wave jerks) :固视时出现的不自主冲动性眼球运动。
平滑追踪障碍 :视标追踪呈阶梯状(追踪异常)。
扫视 辨距不良(saccadic dysmetria)扫视 。
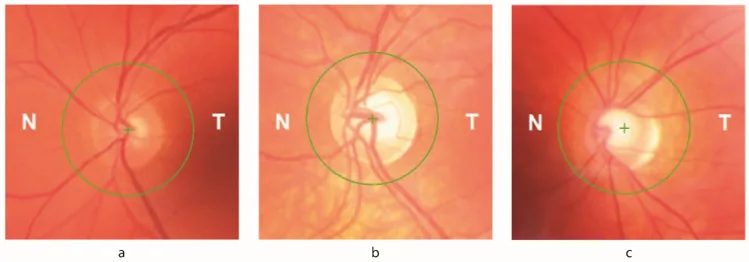
视神经萎缩 眼底检查 可确认。高达30%的患者出现眼科体征。
RNFL 厚度减少OCT 检测。与视力 、对比敏感度 下降直接相关。
视野缺损 视野缺损 。与OCT 记录的RNFL 消失相关。
Q
眼科症状是否必然出现?
A
FRDA中最多30%的患者出现眼科体征,但许多患者早期缺乏视觉自觉症状。视神经萎缩 、RNFL 厚度减少、眼球运动障碍 可通过客观检查检测,并与视力 、对比敏感度 下降直接相关。建议定期进行眼科检查。
FRDA的病因是第9号染色体上FXN(frataxin)基因内含子1中GAA三核苷酸重复序列的扩增。96%的病例为双等位基因致病性扩增的纯合子1) 。其余病例为GAA扩增与点突变,或GAA扩增与基因内/全基因缺失的复合杂合子1) 。
GAA重复次数与疾病的关系如下所示。
分类 GAA重复次数 正常 5~33次 中间(相当于携带者) 34~65次 致病 66次以上(另有报告为90次以上)1) 3)
GAA重复次数越多,发病年龄越早,病情越重。等位基因重复次数极长(如999/766)的病例在11岁发病,5年内需要轮椅(比通常的10年更快)3) 。重复序列中的中断可能延迟发病年龄1) 。
基因内缺失合并GAA扩增的复合杂合子比双等位基因扩增更倾向于早发、快速进展和严重心肌病,但也有报告显示典型病程的病例1) 。
近亲结婚因常染色体隐性遗传 的疾病特性,会增加发病风险2) 3) 。在欧洲血统人群中频率较高。
Q
如果基因检测判定为纯合子,是否可能存在复合杂合子?
A
由于PCR片段分析和TP-PCR无法检测基因内缺失,因此表观上的双等位基因扩增实际上可能是GAA扩增加上基因内缺失1) 。通过MLPA(多重连接依赖性探针扩增)进行额外检测以及亲本样本检测对于准确的遗传咨询 至关重要。
临床诊断采用Harding标准。
类别 主要项目 必需项目 25岁前发病、进行性步态及四肢共济失调、膝腱及跟腱反射消失、轴索变性表现、构音障碍(发病5年后) 附加表现(≥66%) 脊柱侧弯、下肢锥体束性肌力下降、上肢反射消失、粗大神经纤维感觉消失、心电图异常 其他(<50%) 眼震 、视神经萎缩 、听力下降、远端肌萎缩、弓形足、糖尿病
PCR + TP-PCR :扩增内含子1的GAA重复序列,检测延长的等位基因。MLPA(多重连接依赖性探针扩增) :用于检测片段分析或TP-PCR无法发现的缺失/重复1) 。需额外进行以排除表观双等位基因扩增实际为复合杂合子的可能性。Southern blot分析 :与PCR联合使用可实现99%以上的诊断准确率3) 。亲本样本检测 :对准确的遗传咨询 至关重要1) 。
表现为感觉神经病,特征为感觉神经动作电位(SNAP)消失。轴索性感觉多发性神经病的模式具有特征性3) 。
眼电图 (EOG )扫视 (潜伏期和振幅)、追踪(对不同目标速度的增益)、前庭眼反射(对不同头部角速度的增益)以及固视(眼震 波形分析、方波潜伏期)。光学相干断层扫描 (OCT )视网膜神经纤维层 (RNFL )厚度。可确认与视力 和对比敏感度 下降的相关性。视野检查 缺损 模式。眼底检查 视神经萎缩 。
以眼球运动障碍 为主诉时,需要与以下疾病进行鉴别。
脊髓小脑变性症(SCD)的散发病例 :包括多系统萎缩的部分症状。Chiari畸形 :以下跳性眼震 导致的视物晃动为特征。遗传性SCD :如SCA 6型、31型、3型等。获得性障碍 :由炎症、肿瘤、血管障碍引起的小脑及脑干病变。
心电图与超声心动图 :评估心肌病、左室壁厚度、射血分数及舒张功能2) 。需要定期监测。血糖相关检查 2) :通过空腹血糖、HbA1c、C肽、空腹胰岛素、胰岛自身抗体(GAD65、IA-2、ZnT8)、HOMA2-IR、HOMA2-%B进行糖尿病的早期发现和病型鉴别。临床评估量表 :使用FARS(Friedreich共济失调评定量表)、ICARS、SARA对神经症状进行定量评估2) 。SARA显示自然病程每年恶化0.77分(标准误0.06)2) 。脑和脊髓MRI :评估小脑萎缩(有发病3年后出现的病例3) )和脊髓变性表现。
目前尚无根治性治疗,多学科协作的对症治疗和并发症管理是治疗的核心。
神经内科·骨科
脊柱侧弯管理 :轻至中度采用支具治疗,重度考虑手术。
凹足管理 :通过腓肠肌注射肉毒毒素、跟腱拉伸改善活动能力。
康复治疗 :通过物理治疗、作业治疗、语言治疗维持功能。
眼科
低视力 护理 :对视神经萎缩 、视网膜 萎缩导致视觉症状的患者,提供放大镜、照明调整及生活指导。
眼球运动障碍 的对症治疗眼震 ,使用棱镜眼镜 (在加重的眼位方向上,对双眼附加相同棱镜度数)。对于垂直眼震 、周期性方向交替性眼震 、冲动性眼球运动混入,给予GABA_B受体激动剂。
处方示例(GABA_B受体激动剂) :加巴喷丁片(5 mg)3~6片,分1~3次服用。
FRDA相关的糖尿病由于线粒体功能障碍为根本原因,药物选择需注意2) 。
应避免的药物 :二甲双胍和噻唑烷二酮类药物因抑制线粒体复合体I而应避免使用2) 。磺脲类药物存在β细胞应激和低血糖风险。推荐的降糖药物 :DPP-4抑制剂(如西格列汀100 mg/日)和GLP-1类似物为首选2) 。伊美格列明(500 mg×2/日)作为一种靶向线粒体功能的新型降糖药可能有用2) 。胰岛素治疗 :以0.5 U/kg/日为目标起始2) 。线粒体靶向辅助治疗 :有报道联合使用L-肉碱500 mg/日、辅酶Q10 100 mg/日和维生素E 400 IU/日2) 。神经营养维生素 :维生素B1 10 mg、B2 10 mg、B3 45 mg、B5 50 mg、B6 3 mg、B12 15 mcg/日2) 。
使用上述方案的病例报告显示,HbA1c从13.3%降至8.4%(17个月后),进一步降至6.9%(再19个月后),ICARS从85改善至71,改善14分2) 。
尚无根治方法,神经症状呈进行性发展。
FRDA相关的糖尿病需要选择与普通2型糖尿病不同的药物。应避免使用抑制线粒体复合体I的二甲双胍和噻唑烷二酮类药物2) 。
心肌病是死亡的主要原因,定期心脏评估(心电图、超声心动图)必不可少。
Q
弗里德赖希共济失调相关的糖尿病适合哪种治疗?
A
由于存在线粒体功能障碍,应避免使用二甲双胍和噻唑烷二酮类药物2) 。推荐使用DPP-4抑制剂(如西格列汀)和GLP-1类似物。L-肉碱、辅酶Q10和维生素E等线粒体靶向辅助治疗也可能有用2) 。
frataxin是一种定位于线粒体内膜的蛋白质。它参与铁代谢(铁储存、铁硫簇组装),对线粒体呼吸链的正常功能至关重要1) 。
GAA重复序列的延长导致转录沉默,降低FXN mRNA水平1) 。frataxin缺乏会导致以下情况。
线粒体内铁蓄积 :由于铁处理功能下降所致。ATP生成减少 :呼吸链复合体功能障碍2) 。ROS增加 :氧化应激 增强1) 2) 。
后根神经节 :萎缩变小,后根变细呈灰色。脊髓 :整体直径缩小,尤其在胸髓区域明显。皮质脊髓束、脊髓小脑束和后索发生进行性变性。
由于frataxin缺乏,细胞内铁水平波动,视网膜神经节细胞 (RGC )对氧化应激 变得脆弱。高达30%的FA 患者出现眼科体征。脑干和小脑回路以及视神经 均受累。RNFL 厚度减少与视力 和对比敏感度 下降直接相关,从周边向中心同心圆状进展的早期视野缺损 与OCT 记录的RNFL 消失相关。
肥厚型心肌病导致心脏重量增加。心肌组织肉眼可见呈“大理石样”外观。
线粒体功能障碍是FRDA相关糖尿病的根本原因2) 。
β细胞功能障碍 :ATP生成障碍导致胰岛素分泌减少,最终导致β细胞丢失。α细胞分泌调节异常 :高血糖时出现反常的高胰高血糖素血症,低血糖时胰高血糖素反应不足2) 。胰岛素抵抗的机制 :骨骼肌氧化磷酸化障碍导致葡萄糖摄取减少,脂质代谢障碍导致肝脏和肌肉异位脂肪沉积,慢性炎症和氧化应激 导致胰岛素信号障碍,自主神经功能障碍导致肝脏葡萄糖生成异常,共济失调导致身体不活动和肌肉萎缩,继发性代谢恶化2) 。
携带基因内缺失的患者比双等位基因扩增患者更倾向于早发、快速进展和严重心肌病1) 。如果缺失导致起始密码子被移除,则蛋白质生成完全消失1) 。
Q
为什么弗里德赖希共济失调会出现眼部症状?
A
共济蛋白缺乏导致的铁代谢异常和氧化应激 会损伤视网膜神经节细胞 。RNFL 厚度减少与视力 和对比敏感度 下降直接相关,并导致从周边向中心同心圆状进展的视野缺损 。此外,脑干和小脑回路受损会引起眼球震颤 、追踪眼球运动障碍 和扫视 测量障碍等眼球运动异常。
Cooper等人(2008)的临床试验报告称,CoQ10与维生素E联合治疗两年后,ICARS评分显著改善2) 。
Schöls等人(2005)的随机安慰剂对照交叉试验显示,L-肉碱给药可显著改善线粒体ATP生成2) 。
Sureshkumar等人(2025)对一名患有FA 相关糖尿病的32岁女性患者,联合使用了胰岛素疗法、西格列汀、L-肉碱、CoQ10、维生素E、神经营养维生素和伊美格列明2) 。HbA1c从13.3%在17个月后降至8.4%,19个月后进一步降至6.9%;ICARS评分从85降至71,改善14分(HOMA2-IR:4.5→1.2,HOMA2-%B:5→60,MAGE:120→70 mg/dL,CV:43%→34.9%)。根据自然病程,SARA评分预计3年内恶化约2.31分,而该病例显示出约16分的正向偏差。这被认为是首次报告长期血糖稳定与共济失调改善同时实现的病例。
伊美格列明被认为通过改善呼吸链活性、减轻氧化应激 、促进ATP/NAD+合成发挥作用2) 。
AAV载体疗法 :AAVrh.10hFXN旨在将正常FXN基因导入心脏和神经系统的临床试验正在进行中,已显示FXN表达和疾病标志物的早期改善(Munoz-Zuluaga et al. 2023)2) 。CRISPR-Cas9 :旨在去除GAA重复序列,正在YG8R来源细胞和小鼠模型中进行研究(Ouellet et al. 2017)2) 。挑战 :需要控制免疫反应并建立安全的基因递送方法2) 。
Aguilera等人(2023)通过对疑似双等位基因扩增患者的亲本样本检测,鉴定出FXN基因5’UTR和外显子1-2包含的新型基因内缺失1) 。尽管文献中仅报告了10例基因内缺失,但实际频率可能更高。使用MLPA等补充基因分析和亲本样本检测的标准化是未来的课题。
西非首次确诊病例的报告表明,需要在不同民族和地理背景下进行大规模队列研究3) 。疾病修饰变异的鉴定可能成为未来的治疗靶点。
Aguilera C, Esteve-Garcia A, Casasnovas C, et al. Novel intragenic deletion within the FXN gene in a patient with typical phenotype of Friedreich ataxia: may be more prevalent than we think? BMC Med Genomics. 2023;16:312.
Sureshkumar P, Kumar SS, Cheriyan J, Masood A. Friedreich Ataxia and Related Diabetes: Therapeutic Approach Targeting Mitochondrial Dysfunction. JCEM Case Rep. 2025;3:luaf215.
Cissé CAK, Cissé L, Ba HO, et al. Friedreich ataxia in a family from Mali, West Africa. Clin Case Rep. 2021;9:e04065.
复制全文后,可以粘贴到你常用的 AI 助手中提问。
打开下面的 AI 助手,并把复制的内容粘贴到聊天框。