L’ataxie de Friedreich (FRDA) est une maladie génétique progressive affectant les systèmes nerveux central et périphérique. C’est l’ataxie récessive autosomique la plus fréquente dans la population blanche.
En 1863, Nikolaus Friedreich a rapporté une ataxie juvénile, une scoliose et une dégénérescence cardiaque familiale. Plus tard, Pierre Marie a distingué l’ataxie de Friedreich des autres ataxies, établissant ainsi le concept de la maladie.
Sur le plan épidémiologique, il existe de grandes variations régionales, avec une prévalence rapportée de 1:20 000 à 1:750 000. Chez les Européens, elle est d’environ 1:21 0001), et dans le monde entier, on estime qu’elle est de 1:40 0002) à 1:50 0003). La fréquence des porteurs est d’environ 1/701). La fréquence est élevée dans le sud de la France, le nord de l’Espagne et l’Irlande, et faible en Scandinavie et en Russie. Elle est encore plus faible en Afrique subsaharienne et dans l’hémisphère oriental, mais des cas confirmés génétiquement ont été rapportés en Afrique de l’Ouest (dans une famille touarègue consanguine au Mali)3).
Le mode de transmission est autosomique récessif, et le taux d’incidence est égal chez les hommes et les femmes. L’âge typique d’apparition est en moyenne de 15,5 ans, et la plupart des cas surviennent avant 25 ans. L’apparition est fréquente entre 8 et 15 ans2). L’espérance de vie moyenne est de 39 ans, et la principale cause de décès est la cardiomyopathie2).
QÀ quelle fréquence survient l'ataxie de Friedreich ?
A
Il s’agit de l’ataxie héréditaire la plus fréquente dans la population blanche, avec une prévalence variant de 1:20 000 à 1:750 000 selon les régions. En Europe, elle touche environ 1 personne sur 21 0001), et dans le monde, on estime qu’elle touche 1 personne sur 40 000 à 50 0002)3). La fréquence des porteurs est d’environ 1/701).
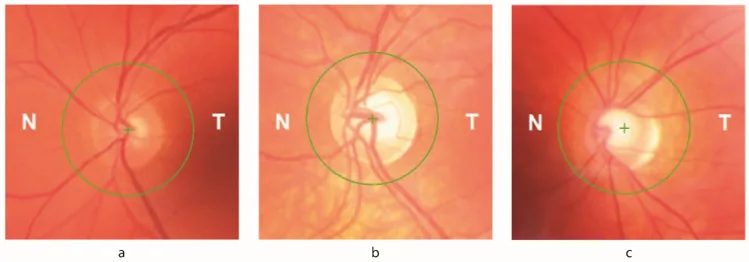
Petya Bogdanova-Mihaylova, Helena Maria Plapp, Hongying Chen et al. Longitudinal Assessment Using Optical Coherence Tomography in Patients with Friedreich’s Ataxia. Tomography. 2021 Dec 8; 7(4):915. Figure 1. PMCID: PMC8706975. License: CC BY.
Troubles de la marche : symptôme initial le plus fréquent. Commence par une démarche instable dans toutes les directions.
Maladresse et tremblements des membres supérieurs : certains cas débutent par un tremblement des mains3).
Dysarthrie : apparaît souvent dans les 10 à 15 ans suivant le début de la maladie.
Symptômes sensoriels : perte de la sensibilité profonde (proprioception).
Symptômes visuels : certains patients présentent une baisse de l’acuité visuelle et de la sensibilité au contraste, mais la majorité ne présente aucun symptôme visuel subjectif au stade précoce.
La progression des symptômes moteurs varie selon les individus, mais en moyenne, la marche autonome devient impossible environ 8 ans après l’apparition des premiers symptômes, et un fauteuil roulant est nécessaire après 11 à 15 ans. Dans les cas où le nombre de répétitions GAA est élevé, il a été rapporté que le fauteuil roulant était nécessaire après 5 ans 3).
Signes cliniques (constatations du médecin lors de l’examen)
Démarche ataxique : instabilité de la marche dans toutes les directions. Accompagnée de dysmétrie, hypotonie, dysdiadococinésie et trouble de la coordination temporelle des mouvements1).
Abolition des réflexes ostéotendineux profonds : L’abolition des réflexes rotulien et achilléen est un critère obligatoire dans les critères diagnostiques de Harding. L’abolition des réflexes des membres supérieurs est également fréquente. Certains cas présentent une hyperréflexie, une spasticité ou une démarche en ciseaux3).
Signe de Babinski : Apparaît comme une réponse plantaire en extension.
Nystagmus : Dû à une atteinte des circuits cérébelleux et du tronc cérébral. Comprend un nystagmus dépendant de la position du regard.
Saccades en créneaux (square-wave jerks) : Mouvements oculaires involontaires et impulsifs survenant pendant la fixation.
Trouble de la poursuite oculaire : La poursuite d’une cible devient saccadée (anomalie de poursuite).
Dysmétrie saccadique (saccadic dysmetria) : Apparition de saccades trop amples ou trop courtes.
Atrophie optique : Observée à l’examen du fond d’œil. Des signes ophtalmologiques apparaissent chez jusqu’à 30 % des patients.
Diminution de l’épaisseur de la couche des fibres nerveuses rétiniennes (RNFL) : Détectée par OCT. Corrélée directement à une baisse de l’acuité visuelle et de la sensibilité au contraste.
Déficit du champ visuel : Déficit visuel initial progressant de manière concentrique depuis la périphérie. Associé à la perte de RNFL enregistrée par OCT.
QLes symptômes oculaires apparaissent-ils toujours ?
A
Dans la FRDA, jusqu’à 30 % des patients présentent des signes ophtalmologiques, mais la plupart n’ont pas de symptômes visuels subjectifs au début. L’atrophie optique, la diminution de l’épaisseur de la couche de fibres nerveuses rétiniennes et les troubles des mouvements oculaires sont détectés par des examens objectifs et sont directement corrélés à une baisse de l’acuité visuelle et de la sensibilité au contraste. Un examen ophtalmologique régulier est recommandé.
La cause de la FRDA est l’expansion de la répétition de trois nucléotides GAA dans l’intron 1 du gène FXN (frataxine) sur le chromosome 9. 96% des cas sont homozygotes pour une expansion pathogène biallélique1). Les cas restants sont des hétérozygotes composites avec une expansion GAA et une mutation ponctuelle, ou une expansion GAA et une délétion intragénique/de gène entier1).
La relation entre le nombre de répétitions GAA et la maladie est présentée ci-dessous.
Classification
Nombre de répétitions GAA
Normal
5 à 33
Intermédiaire (porteur)
34 à 65
Pathologique
66 répétitions ou plus (90 répétitions ou plus selon d’autres rapports) 1)3)
Plus le nombre de répétitions GAA est élevé, plus l’âge d’apparition est précoce et plus la maladie est sévère. Chez un patient avec un nombre de répétitions alléliques extrêmement élevé de 999/766, la maladie s’est déclarée à 11 ans et un fauteuil roulant a été nécessaire en moins de 5 ans (plus rapide que les 10 ans habituels) 3). Les interruptions dans la séquence répétée peuvent retarder l’âge d’apparition 1).
Les hétérozygotes composites avec délétion intragénique + expansion GAA auraient tendance à avoir un début plus précoce, une progression plus rapide et une cardiomyopathie plus sévère que les expansions bialléliques, mais des cas avec une évolution typique ont également été rapportés 1).
Les mariages consanguins augmentent le risque de développer la maladie en raison de son mode de transmission autosomique récessif2)3). Cette pathologie est plus fréquente dans les populations d’ascendance européenne.
QSi le test génétique montre un statut homozygote, est-il possible qu'il s'agisse en réalité d'un hétérozygote composite ?
A
L’analyse par PCR des fragments et la TP-PCR ne détectent pas les délétions intragéniques. Ainsi, une expansion biallélique apparente peut en réalité correspondre à une expansion GAA associée à une délétion intragénique1). Des tests complémentaires par MLPA (Multiplex Ligation-dependent Probe Amplification) et l’analyse des échantillons parentaux sont indispensables pour un conseil génétique précis.
Les critères de Harding sont utilisés pour le diagnostic clinique.
Catégorie
Principaux éléments
Critères obligatoires
Début avant 25 ans, ataxie progressive de la marche et des membres, abolition des réflexes rotuliens et achilléens, neuropathie axonale, dysarthrie (après 5 ans d’évolution)
Signes supplémentaires (≥66%)
Scoliose, faiblesse pyramidale des membres inférieurs, aréflexie des membres supérieurs, perte sensorielle des fibres nerveuses épaisses, anomalies ECG
PCR + TP-PCR : Amplification de la répétition GAA dans l’intron 1 pour détecter l’allèle expansé.
MLPA (Multiplex Ligation-dependent Probe Amplification) : nécessaire pour diagnostiquer les délétions et duplications non détectables par analyse de fragments ou TP-PCR1). Ajouté pour exclure la possibilité qu’une expansion biallélique apparente soit un hétérozygote composite.
Analyse par Southern blot : combinée à la PCR, elle permet un diagnostic avec une précision supérieure à 99 %3).
Examen des échantillons parentaux : indispensable pour un conseil génétique précis1).
Montre une neuropathie sensorielle caractérisée par une disparition du potentiel d’action nerveux sensitif (SNAP). Le profil d’une neuropathie sensitive axonale multifocale est caractéristique3).
Électro-oculographie (EOG) : utilisée pour l’analyse qualitative et quantitative des mouvements oculaires. Évalue les saccades visuellement guidées (latence et amplitude), la poursuite (gain pour différentes vitesses de cible), le VOR (gain pour différentes vitesses angulaires de la tête) et la fixation (analyse de la forme du nystagmus, latence des ondes carrées).
Tomographie par cohérence optique (OCT) : mesure quantitative de l’épaisseur de la couche des fibres nerveuses rétiniennes (RNFL). Permet de confirmer la corrélation avec la diminution de l’acuité visuelle et de la sensibilité au contraste.
Examen du champ visuel : évaluer le motif de déficit qui progresse de manière concentrique depuis la périphérie.
Examen du fond d’œil : vérifier la présence d’une atrophie du nerf optique.
Électrocardiogramme et échocardiographie : évaluent la cardiomyopathie, l’épaisseur de la paroi ventriculaire gauche, la fraction d’éjection et la fonction diastolique2). Une surveillance régulière est nécessaire.
Tests liés à la glycémie2) : glycémie à jeun, HbA1c, peptide C, insuline à jeun, auto-anticorps des îlots pancréatiques (GAD65, IA-2, ZnT8), HOMA2-IR, HOMA2-%B pour la détection précoce et la classification du diabète.
Échelles d’évaluation clinique : FARS (Friedreich Ataxia Rating Scale), ICARS, SARA pour l’évaluation quantitative des symptômes neurologiques2). Avec SARA, une aggravation annuelle de 0,77 point (SE 0,06) a été rapportée dans l’évolution naturelle2).
IRM cérébrale et médullaire : évaluation de l’atrophie cérébelleuse (apparue 3 ans après le début dans certains cas3)) et des signes de dégénérescence médullaire.
Actuellement, il n’existe pas de traitement curatif ; la prise en charge repose sur un traitement symptomatique multidisciplinaire et la gestion des complications.
Gestion de la scoliose : pour les cas légers à modérés, traitement par corset ; pour les cas sévères, envisager une chirurgie.
Gestion du pied creux : injection de toxine botulique dans le muscle gastrocnémien et étirement du tendon d’Achille pour améliorer la mobilité.
Réadaptation : maintien des fonctions par kinésithérapie, ergothérapie et orthophonie.
Ophtalmologie
Soins de basse vision : pour les patients présentant des symptômes visuels dus à une atrophie du nerf optique ou de la rétine, fournir des loupes, ajuster l’éclairage et donner des conseils sur les activités quotidiennes.
Traitement symptomatique des troubles oculomoteurs : Pour le nystagmus dépendant de la position des yeux, on utilise des verres prismatiques (en ajoutant la même puissance de prisme aux deux yeux dans la direction qui aggrave la position). Pour le nystagmus vertical, le nystagmus alternant périodique et les mouvements oculaires saccadés, on administre un agoniste GABA_B.
Exemple de prescription (agoniste GABA_B) : 3 à 6 comprimés de Gabaron (5 mg), en 1 à 3 prises.
Le diabète associé à la FRDA étant dû à un dysfonctionnement mitochondrial, la sélection des médicaments nécessite une attention particulière2).
Médicaments à éviter : La metformine et les thiazolidinediones inhibent le complexe I mitochondrial, donc à éviter2). Les sulfonylurées présentent un risque de stress des cellules bêta et d’hypoglycémie.
Médicaments hypoglycémiants recommandés : Les inhibiteurs de la DPP-4 (ex. sitagliptine 100 mg/jour) et les analogues du GLP-1 sont préférés2). L’iméglimine (500 mg x 2/jour) pourrait être utile comme nouvel hypoglycémiant ciblant la fonction mitochondriale2).
Insulinothérapie : commencer à environ 0,5 U/kg/jour2).
Thérapie adjuvante ciblant les mitochondries : l’association de L-carnitine 500 mg/jour, CoQ10 100 mg/jour et vitamine E 400 UI/jour a été rapportée2).
Dans le rapport de cas utilisant le régime ci-dessus, l’HbA1c s’est améliorée de 13,3 % à 8,4 % (après 17 mois), puis à 6,9 % (après 19 mois supplémentaires), et l’ICARS a diminué de 85 à 71, soit une amélioration de 14 points2).
QQuel traitement est approprié pour le diabète associé à l'ataxie de Friedreich ?
A
En raison du dysfonctionnement mitochondrial sous-jacent, la metformine et les thiazolidinediones doivent être évitées2). Les inhibiteurs de la DPP-4 (comme la sitagliptine) et les analogues du GLP-1 sont recommandés. Les thérapies adjuvantes ciblant les mitochondries, telles que la L-carnitine, le CoQ10 et la vitamine E, peuvent également être utiles2).
6. Physiopathologie et mécanisme détaillé de la maladie
La frataxine est une protéine localisée dans la membrane interne des mitochondries. Elle est impliquée dans le métabolisme du fer (stockage du fer et assemblage des clusters fer-soufre) et est essentielle au fonctionnement normal de la chaîne respiratoire mitochondriale1).
L’expansion des répétitions GAA provoque un silençage transcriptionnel (transcriptional silencing) et réduit les niveaux d’ARNm de FXN1). La déficience en frataxine entraîne ce qui suit.
Accumulation de fer mitochondrial : due à une altération du traitement du fer.
Diminution de la production d’ATP : dysfonctionnement des complexes de la chaîne respiratoire2).
Ganglion spinal : petit et atrophié, la racine postérieure est fine et de couleur grise.
Moelle épinière : réduction du diamètre sur toute sa longueur, particulièrement marquée dans la région thoracique. Dégénérescence progressive des voies corticospinales, spinocérébelleuses et des cordons postérieurs.
La carence en frataxine entraîne des variations des niveaux de fer intracellulaire, rendant les cellules ganglionnaires rétiniennes (CGR) vulnérables au stress oxydatif. Jusqu’à 30 % des patients atteints de FA présentent des signes ophtalmiques. Les circuits du tronc cérébral et du cervelet ainsi que le nerf optique sont tous deux affectés. La diminution de l’épaisseur de la RNFL est directement corrélée à une baisse de l’acuité visuelle et de la sensibilité au contraste, et les déficits du champ visuel initial, qui progressent de manière concentrique depuis la périphérie, sont associés à une perte de la RNFL documentée par OCT.
L’hypertrophie cardiaque due à la cardiomyopathie hypertrophique augmente le poids du cœur. Le tissu myocardique peut présenter un aspect macroscopique « marbré » (marble-like).
Le dysfonctionnement mitochondrial est la cause sous-jacente du diabète associé à la FRDA2).
Dysfonctionnement des cellules β : la production altérée d’ATP réduit la sécrétion d’insuline, conduisant finalement à la perte des cellules β.
Dysrégulation de la sécrétion des cellules α : hyperglucagonémie paradoxale en hyperglycémie et réponse glucagonique insuffisante en hypoglycémie2).
Mécanismes de l’insulinorésistance : diminution de la captation du glucose due à une altération de la phosphorylation oxydative dans le muscle squelettique, accumulation ectopique de graisse dans le foie et les muscles due à un trouble du métabolisme lipidique, altération de la signalisation insulinique due à l’inflammation chronique et au stress oxydatif, production hépatique anormale de glucose due à la neuropathie autonome, et détérioration métabolique secondaire due à l’inactivité physique et à l’atrophie musculaire liées à l’ataxie2).
Les porteurs de délétion intragénique ont tendance à présenter un début plus précoce, une progression rapide et une cardiomyopathie sévère par rapport à l’expansion biallélique1). Si la délétion supprime le codon start, la production de protéines est complètement abolie1).
QPourquoi l'ataxie de Friedreich provoque-t-elle des symptômes oculaires ?
A
Le métabolisme anormal du fer et le stress oxydatif dus à la carence en frataxine endommagent les cellules ganglionnaires de la rétine. La diminution de l’épaisseur de la RNFL est directement corrélée à une baisse de l’acuité visuelle et de la sensibilité au contraste, entraînant un déficit du champ visuel qui progresse de manière concentrique depuis la périphérie. De plus, les lésions des circuits du tronc cérébral et du cervelet provoquent des anomalies des mouvements oculaires telles que le nystagmus, les troubles de la poursuite oculaire et les troubles de la mesure des saccades.
7. Recherches récentes et perspectives futures (rapports en phase de recherche)
L’essai clinique de Cooper et al. (2008) a rapporté une amélioration significative du score ICARS sur deux ans grâce à une thérapie combinée de CoQ10 et de vitamine E2).
L’essai croisé randomisé contrôlé par placebo de Schöls et al. (2005) a montré que l’administration de L-carnitine améliorait significativement la production d’ATP mitochondriale2).
Sureshkumar et al. (2025) ont traité une femme de 32 ans atteinte de diabète lié à l’ataxie de Friedreich avec une combinaison d’insulinothérapie, de sitagliptine, de L-carnitine, de CoQ10, de vitamine E, de vitamines neurotropes et d’iméglimine2). L’HbA1c est passée de 13,3 % à 8,4 % après 17 mois, puis à 6,9 % après 19 mois, et l’ICARS a diminué de 85 à 71, soit une amélioration de 14 points (HOMA2-IR : 4,5→1,2 ; HOMA2-%B : 5→60 ; MAGE : 120→70 mg/dL ; CV : 43 %→34,9 %). Alors que l’histoire naturelle selon SARA prédit une aggravation d’environ 2,31 points sur trois ans, un écart positif d’environ 16 points a été observé. Il s’agit du premier cas rapporté d’une stabilisation glycémique à long terme et d’une amélioration de l’ataxie obtenues simultanément.
On pense que l’iméglimine agit en améliorant l’activité de la chaîne respiratoire, en réduisant le stress oxydatif et en favorisant la synthèse d’ATP/NAD+2).
Thérapie par vecteur AAV : L’essai clinique AAVrh.10hFXN, visant à introduire le gène FXN normal dans le cœur et le système nerveux, est en cours et a montré une amélioration précoce de l’expression de FXN et des marqueurs de la maladie (Munoz-Zuluaga et al. 2023)2).
CRISPR-Cas9 : visant à éliminer les séquences répétées GAA, des études sont en cours sur des cellules et modèles murins YG8R (Ouellet et al. 2017)2).
Défi : Il est nécessaire de contrôler la réponse immunitaire et d’établir une méthode sûre de délivrance de gènes2).
Amélioration de la précision du diagnostic génétique
Aguilera et al. (2023) ont identifié une nouvelle délétion intragénique incluant la région 5’UTR et les exons 1-2 du gène FXN en testant des échantillons parentaux d’un patient présumé porteur d’une expansion biallélique 1). Seulement 10 délétions intragéniques ont été rapportées dans la littérature jusqu’à présent, mais il est suggéré qu’elles pourraient être plus fréquentes. La standardisation des analyses génétiques complémentaires utilisant la MLPA et des tests parentaux est considérée comme un défi futur.
Le signalement du premier cas confirmé en Afrique de l’Ouest souligne la nécessité d’études de cohorte à grande échelle dans divers contextes ethniques et géographiques3). L’identification de variants modificateurs de la maladie pourrait constituer des cibles thérapeutiques futures.
Aguilera C, Esteve-Garcia A, Casasnovas C, et al. Novel intragenic deletion within the FXN gene in a patient with typical phenotype of Friedreich ataxia: may be more prevalent than we think? BMC Med Genomics. 2023;16:312.
Sureshkumar P, Kumar SS, Cheriyan J, Masood A. Friedreich Ataxia and Related Diabetes: Therapeutic Approach Targeting Mitochondrial Dysfunction. JCEM Case Rep. 2025;3:luaf215.
Cissé CAK, Cissé L, Ba HO, et al. Friedreich ataxia in a family from Mali, West Africa. Clin Case Rep. 2021;9:e04065.
Copiez le texte de l'article et collez-le dans l'assistant IA de votre choix.
Article copié dans le presse-papiers
Ouvrez un assistant IA ci-dessous et collez le texte copié dans la conversation.