Die akute exsudative polymorphe vitelliforme Makulopathie (Acute Exudative Polymorphous Vitelliform Maculopathy; AEPVM) ist eine seltene Netzhauterkrankung, die durch bilaterale, multiple, gelb-weiße subretinale Läsionen in Übereinstimmung mit einer serösen Netzhautablösung gekennzeichnet ist. Sie wurde erstmals 1988 von Gass et al. beschrieben (Trans Am Ophthalmol Soc 1988).
Das Erkrankungsalter reicht von 13 bis 69 Jahren, ohne Geschlechtsunterschied. 1) Sie tritt hauptsächlich bei Weißen auf, wurde aber auch bei anderen Ethnien berichtet. Die idiopathischen Fälle in der Literatur sind mit etwa 20 extrem selten. 1)
Die AEPVM wird nach der Ätiologie in folgende zwei Typen eingeteilt:
Idiopathisch: Es wird angenommen, dass eine Virusinfektion oder ein Autoimmunmechanismus beteiligt ist.
Paraneoplastisch: Verursacht durch bösartige Tumoren oder Immun-Checkpoint-Inhibitoren.
Diese Erkrankung wird im ICD-10 unter H35.89 (Sonstige näher bezeichnete Netzhautkrankheiten) klassifiziert.
QWie selten ist die AEPVM?
A
In der Literatur wurden nur etwa 20 Fälle idiopathischer AEPVM berichtet, was sie extrem selten macht. 1) Unter Einbeziehung der paraneoplastischen Form steigt die Zahl der Berichte, aber beide Typen sind weltweit selten.
Der wichtigste Fundusbefund sind beidseitige, symmetrische, multiple gelb-weiße subretinale Läsionen. Die Morphologie der Läsionen ist vielfältig: rund, oval, kurvilinear usw. Sie befinden sich hauptsächlich in der Makularegion, aber auch bläschenartige (bleb-like) Läsionen entlang der Gefäßbögen können auftreten. 1)
Das vitelliforme Material in den Läsionen kann sich durch die Schwerkraft nach unten absetzen und einen meniskusartigen Flüssigkeitsspiegel (Pseudohypopyon) bilden. 1) Es gibt keine Glaskörperentzündung oder Vaskulitiszeichen, und die Papille ist unauffällig. 1)
Die Fundusbefunde unterscheiden sich zwischen der akuten Phase und der Erholungsphase. Die beiden Stadien werden im Folgenden beschrieben.
Vergleich zwischen akuter Phase und Erholungsphase:
Die AEPVM wird in zwei Typen unterteilt: idiopathisch und paraneoplastisch, mit jeweils unterschiedlichen Ursachen und Pathomechanismen.
Idiopathisch
Infektionsvorgeschichte: Virale Prodromalsymptome (HCV, Coxsackie B, EBV, HIV, COVID-19), Syphilis, Lyme-Borreliose und andere Infektionen können vorausgehen.
Augentrauma: Ein Augentrauma vor dem Ausbruch wurde als Auslöser berichtet.
Autoimmunhypothese: Anti-Peroxiredoxin-3 (PRDX3)-Antikörper werden in der akuten Phase nachgewiesen und verschwinden nach Remission. Es wird angenommen, dass Autoantikörper gegen RPE- und Photorezeptorproteine an der Pathogenese beteiligt sind.
Fälle ungeklärter Ursache: Es gibt auch rein idiopathische Fälle, bei denen systemische und genetische Untersuchungen negativ sind. 1)
Paraneoplastisch
Melanom: Kutanes Melanom und Aderhautmelanom sind die häufigsten bösartigen Tumoren.
Andere bösartige Tumoren: Assoziationen mit Lungen-, Brust-, Dickdarmkrebs usw. wurden berichtet.
Immun-Checkpoint-Inhibitoren: Die Anwendung von BRAF-Inhibitoren (Vemurafenib, Dabrafenib) oder PD-1-Inhibitoren (Pembrolizumab, Nivolumab) kann mit dem Auftreten verbunden sein.
Entstehungsmechanismus: Es wird angenommen, dass eine Kreuzreaktion (molekulare Mimikry) zwischen Tumorantigenen und Netzhautantigenen eine Autoimmunreaktion gegen RPE und Photorezeptoren auslöst.
Torres-Costa S, Penas S, Carneiro Â, et al. Idiopathic Acute Exudative Polymorphous Vitelliform Maculopathy: Insight into Imaging Features and Outcomes. Case Rep Ophthalmol Med. 2020;2020:7254038. Figure 1 and Figure 2. PMID: 32082665; PMCID: PMC7008265. DOI: 10.1155/2020/7254038. License: CC BY 4.0.
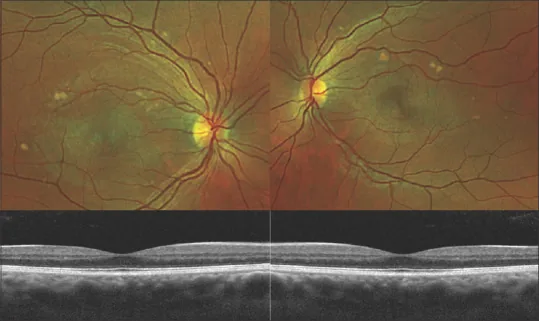
Nebeneinander gestellte Fundusfotos und OCTs beider Augen. Ermöglicht eine übersichtliche Darstellung der Verteilung der Makulaläsionen und der subretinalen Veränderungen.
Die Diagnose einer AEPVM ist eine Ausschlussdiagnose, die eine systematische Abgrenzung ähnlicher Erkrankungen erfordert. Die Kombination multimodaler Bildgebung ist der Schlüssel zur Diagnose. 1)
Bildgebende Untersuchungen
OCT (Optische Kohärenztomographie) : Erkennt eine kuppelförmige neurosensorische Netzhautablösung und hyper- oder isoreflektive subretinale Ablagerungen. In der Erholungsphase kann das Verschwinden der Flüssigkeit und die Wiederanlagerung des RPE beobachtet werden. Auch eine Verdickung der Ellipsoidzone ist feststellbar. 1)
FAF (Autofluoreszenz) : Das vitelliforme Material zeigt eine hohe Autofluoreszenz. Dies ist die wichtigste Untersuchung für die Diagnose. 1) Spiegelt die Ansammlung von Lipofuszin und fluoreszierenden Pigmenten wider.
FA (Fluoreszenzangiographie) : Das vitelliforme Material erscheint hypofluoreszent oder nicht fluoreszierend. Es zeigt ein umgekehrtes Muster im Vergleich zur FAF, was charakteristisch ist. Es werden keine Leckagen festgestellt.
Funktionelle und genetische Untersuchungen
EOG (Elektrookulographie) : Eine verminderte Arden-Ratio wird festgestellt, die auf eine RPE-Dysfunktion hinweist.
Gentests : Die Suche nach BEST1- und PRPH2-Mutationen mittels Next-Generation-Sequencing-Panel ist wichtig. Ein negatives Ergebnis dieser Mutationen kann eine Best’sche vitelliforme Makuladystrophie ausschließen. 1)
Ganzkörperuntersuchung : Eine umfassende Untersuchung zum Ausschluss eines bösartigen Tumors ist obligatorisch. 1)
Das umgekehrte Muster von FA und Fundusautofluoreszenz (FAF) ist ein charakteristischer Befund dieser Erkrankung.
Die AEPVM muss von folgenden Erkrankungen abgegrenzt werden.
Best-Vitelliforme Makuladystrophie (BVMD) : Abgrenzung durch Gentest (BEST1-Mutation). Die AEPVM weist keine BEST1-Mutation auf. 1)
Adult-onset foveomakuläre vitelliforme Dystrophie (AVMD) : Abgrenzung durch Vorhandensein oder Fehlen einer PRPH2-Mutation.
Harada-Krankheit (Vogt-Koyanagi-Harada; VKH) : In der FA zeigen sich eine starke Fluoreszenz der Papille und punktförmige Leckagen. Die AEPVM ist durch das Fehlen von Leckagen abgrenzbar.
QWie unterscheidet man den Morbus Best (vitelliforme Makuladystrophie Best)?
A
Morbus Best ist eine erbliche Erkrankung, die durch eine Mutation im BEST1-Gen verursacht wird und durch Gentests unterschieden werden kann. Bei AEPVM sind Gentests mittels Next-Generation-Sequencing negativ für BEST1- und PRPH2-Mutationen. 1) Auch der Verlauf (akuter oder schleichender Beginn) und die Familienanamnese helfen bei der Differenzialdiagnose.
Für AEPVM gibt es keine etablierte medikamentöse Therapie. Die idiopathische AEPVM ist eine selbstlimitierende Erkrankung, bei der eine spontane Erholung erwartet werden kann.
Beobachtung als Grundlage: Regelmäßige Fundusuntersuchungen und OCT-Überwachung durchführen. Im Bericht von Fernandes et al. wurde nach 6 Monaten eine vollständige Remission erreicht. 1)
Steroidtherapie: Die Wirksamkeit bei idiopathischer AEPVM ist nicht bestätigt und wird derzeit nicht als Standardtherapie empfohlen.
Fälle mit choroidaler Neovaskularisation (CNV): Bei Auftreten einer CNV ist die intravitreale Injektion von Anti-VEGF-Medikamenten indiziert.
Paraneoplastische AEPVM: Bei Verdacht auf einen Zusammenhang mit Immun-Checkpoint-Inhibitoren (BRAF-Inhibitoren, PD-1-Inhibitoren) sollte die Entscheidung über das Absetzen des Medikaments in Absprache mit der Onkologie unter Berücksichtigung des Behandlungsstatus des Primärtumors getroffen werden. Ein Wechsel zu einer alternativen Immuntherapie kann in Betracht gezogen werden.
QWie ist die Sehprognose bei idiopathischer AEPVM?
A
Die idiopathische AEPVM ist eine selbstlimitierende Erkrankung, und in den meisten Fällen ist eine spontane Seherholung zu erwarten. Im Fall von Fernandes et al. verbesserte sich der bestkorrigierte Visus beider Augen nach 6 Monaten auf 20/25, und das OCT bestätigte das vollständige Verschwinden der subretinalen Flüssigkeit. 1) Der Grad der Erholung variiert jedoch von Fall zu Fall.
6. Pathophysiologie und detaillierter Pathomechanismus
Eine Funktionsstörung des retinalen Pigmentepithels (RPE) führt zur Akkumulation von Lipofuszin, was eine seröse Netzhautablösung zur Folge hat. Die physikalische Trennung von neurosensorischer Netzhaut und RPE beeinträchtigt die normale Phagozytose der Photorezeptor-Außensegmente. Abgelöste Photorezeptor-Außensegmente sollen sich in den äußeren Netzhautschichten und im subretinalen Raum ansammeln (Spaide-Hypothese). Die in der FAF beobachtete Hyperautofluoreszenz spiegelt diese Ansammlung von Lipofuszin und fluoreszierenden Pigmenten wider.
Autoantikörper gegen RPE- und Photorezeptor-Proteine werden als pathogenetisch relevant angesehen. Folgende Zielantigene für diese Autoantikörper wurden berichtet.
Recoverin
Transducin-α
Peroxiredoxin 3 (PRDX3) : Anti-PRDX3-Antikörper wurden in der akuten Phase der idiopathischen AEPVM nachgewiesen und verschwanden nach Remission in berichteten Fällen.
Carboanhydrase 2
120-kDa-Photorezeptorprotein
145-kDa-Interphotorezeptor-Retinoid-bindendes Protein
Bei paraneoplastischen Fällen wird angenommen, dass eine Kreuzimmunreaktion (molekulare Mimikry) zwischen Tumorantigenen und gemeinsamen Antigenen von Netzhaut und RPE zur Bildung vitelliformer Läsionen führt.
7. Aktuelle Forschung und Zukunftsperspektiven (Berichte aus der Forschungsphase)
Die Kombination von Farbfundusfotografie, SD-OCT und FAF ermöglicht zunehmend eine klinische Diagnose der AEPVM ohne invasive Untersuchung. 1) Jede Modalität liefert komplementäre Informationen, und insbesondere das inverse Muster von FAF und FA ist der Schlüssel zur Diagnose.
Durch den Einsatz von Next-Generation-Sequencing-Panels können Genmutationen in BEST1, PRPH2 usw. umfassend gesucht werden. Die Standardisierung von Gentests trägt zur Verbesserung der Genauigkeit der Ausschlussdiagnose bei. 1)
Neue Herausforderungen im Zeitalter der Immun-Checkpoint-Inhibitoren
Mit der Verbreitung von BRAF-Inhibitoren (Vemurafenib, Dabrafenib) und PD-1-Inhibitoren (Pembrolizumab, Nivolumab) tritt der Zusammenhang zwischen diesen Medikamenten und paraneoplastischer AEPVM als neue klinische Herausforderung auf. Mit der Verbreitung der Immuntherapie könnte die Zahl der gemeldeten Fälle von paraneoplastischer AEPVM zunehmen.
Notwendigkeit von Fallsammlungen und groß angelegten Studien
In der Literatur gibt es nur etwa 20 Fälle von idiopathischer AEPVM. 1) Zur Aufklärung der Ätiologie und Entwicklung von Behandlungen sind multizentrische Fallsammlungen und groß angelegte prospektive Studien erforderlich.
Fernandes et al. (2023) berichteten über einen Fall von idiopathischer AEPVM bei einem 60-jährigen Mann, bei dem sowohl die systemischen Untersuchungen als auch die Gentests mittels Next-Generation-Sequencing-Panel negativ waren. Nach 6 Monaten verbesserte sich der bestkorrigierte Visus auf 20/25 in beiden Augen, und die SD-OCT bestätigte das vollständige Verschwinden der subretinalen Flüssigkeit. 1)
Joana Silva Fernandes, Pedro Prata Gomes, Pedro Neves, João Pedro Marques. Idiopathic acute exudative polymorphous vitelliform maculopathy: the importance of multimodal imaging, systemic workup and genetic testing. BMJ Case Rep. 2023;16(6):e253969. doi:10.1136/bcr-2022-253969.
Osman M, Mehana O, Eissa M, Zeineldin S, Sinha A. Coronavirus Disease 2019-induced Acute Exudative Polymorphous Vitelliform Maculopathy. Middle East Afr J Ophthalmol. 2022;29(4):235-237. PMID: 38162565.
Lentzsch AM, Dooling V, Wegner I, Di Cristanziano V, Sadda SR, Freund KB, et al. ACUTE EXUDATIVE POLYMORPHOUS VITELLIFORM MACULOPATHY ASSOCIATED WITH PRIMARY EPSTEIN-BARR VIRUS INFECTION. Retin Cases Brief Rep. 2022;16(6):740-746. PMID: 33031214.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.