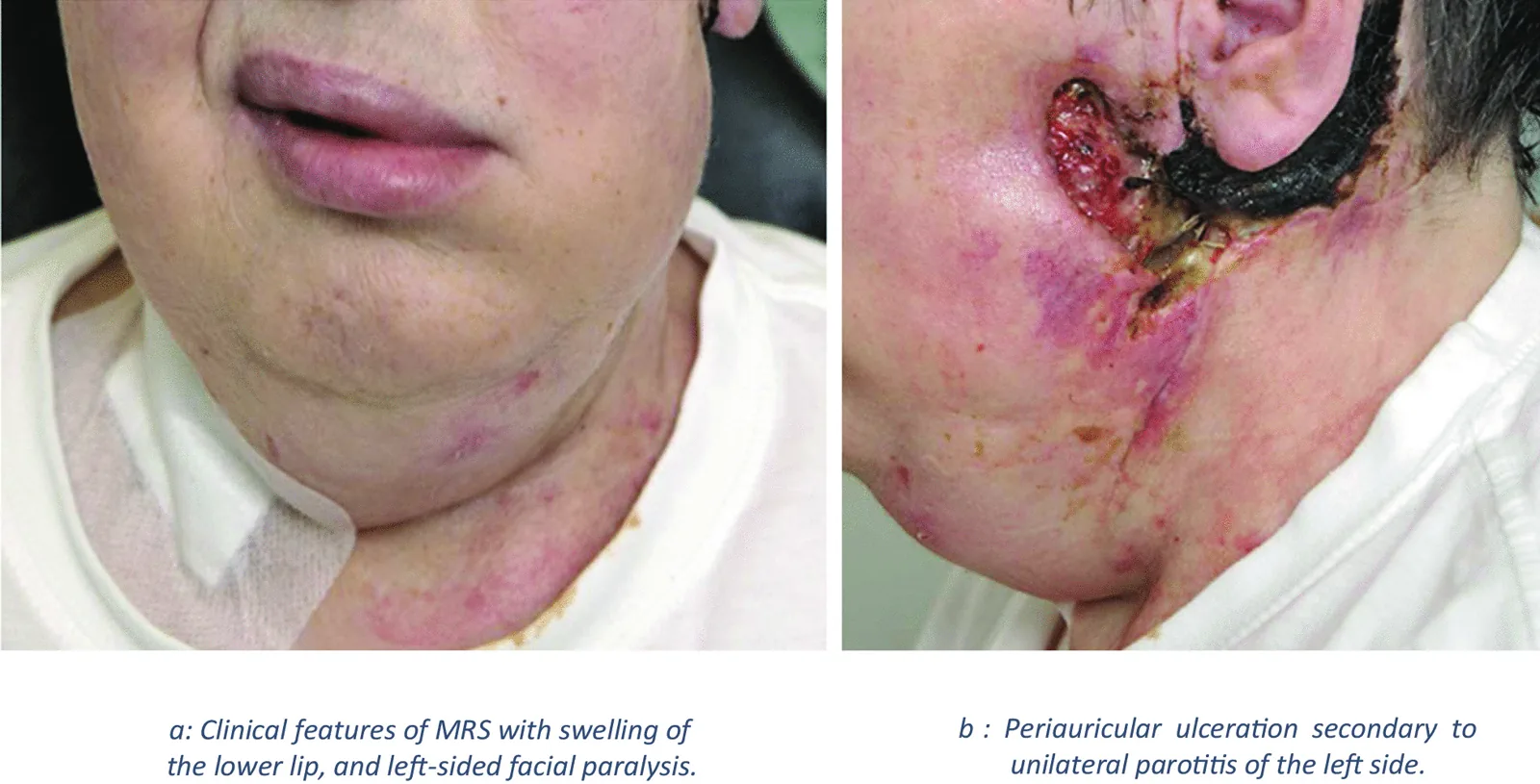
A síndrome de Melkersson-Rosenthal (MRS) é uma doença neurocutânea granulomatosa crônica não caseosa. Apresenta-se com edema oral e facial, e a biópsia revela inflamação granulomatosa. Pertence ao grupo de doenças denominado “granulomatose orofacial”.
O histórico médico desta síndrome é o seguinte. Em 1928, Ernst Melkersson relatou pela primeira vez em detalhes a existência de paralisia facial recorrente e edema. Em 1931, Curt Rosenthal acrescentou a descrição de língua plicada (lingua plicata), e em 1949, Lüscher usou pela primeira vez o nome “Síndrome de Melkersson-Rosenthal” 3).
Epidemiologicamente, estima-se que ocorra em 0,08% da população geral 2). É mais frequente entre os 20 e 30 anos, com idade média de início de 39 anos (variação: 8-79 anos). Predomina em mulheres (cerca de 3 vezes mais).
A tríade clássica consiste nos três seguintes.
Edema orofacial recorrente: especialmente inchaço labial (queilite granulomatosa)
Paralisia facial periférica recorrente
Língua plicata (língua fissurada)
Os três sintomas juntos ocorrem em apenas 8-25% dos casos. A maioria apresenta apenas 1-2 sintomas, e o tipo monossintomático é por vezes descrito como queilite granulomatosa (queilite de Miescher) 1).
Alguns pesquisadores sugerem que esta condição pode ser um dos sintomas da sarcoidose ou doença de Crohn, e permanece incerto se é uma doença independente ou não.
QA síndrome não pode ser diagnosticada se os três sintomas não estiverem todos presentes?
A
A tríade clássica completa está presente em apenas 8–25% dos casos. O diagnóstico é possível se um ou dois sintomas forem confirmados pela história e exame físico, ou se a biópsia mostrar queilite granulomatosa. O diagnóstico definitivo é raro e requer acompanhamento de longo prazo.
J Casper, S Mohammad-Khani, J J Schmidt et al. Melkersson–Rosenthal syndrome in the context of sarcoidosis: a case report . Journal of Medical Case Reports. 2021 Oct 4; 15:488. Figure 2. PMCID: PMC8489098. License: CC BY.
Aspecto clínico de paciente com síndrome de Melkersson-Rosenthal mostrando edema labial
Cada elemento da tríade difere amplamente em frequência e características.
Edema Orofacial
Frequência: Mais alta, 80-100% dos casos.
Características típicas: unilateral, indolor, não compressível. Predileção pelo lábio superior.
Evolução: Inicialmente transitória e intermitente, mas pode tornar-se persistente posteriormente. Pode levar à fibrose tecidual.
Diagnóstico diferencial: Semelhante ao angioedema, mas a MRS é mais persistente e refratária a anti-histamínicos. Níveis de complemento normais.
Paralisia do nervo facial
Frequência de ocorrência: 47-60% dos casos (até 90%). Unilateral ou bilateral.
Evolução: Inicialmente costuma melhorar, mas com recorrências a duração se prolonga e pode tornar-se permanente. Taxa de recorrência de 3-11%.
Relação com edema: Pode aparecer anos antes ou depois do edema orofacial. Em 13-50% dos afetados, edema e paralisia facial ocorrem juntos.
Epidemiologia: A incidência de paralisia facial associada à MRS é de 0,36 por 100.000 pessoas/ano5).
Língua fissurada
Frequência de ocorrência: 30-35% dos casos.
Características: Sulcos com cerca de 2 mm de profundidade e 15 mm de comprimento. Pode ser congênita.
Significado diagnóstico: Também é encontrada em 5% da população normal, portanto, isoladamente não é um critério diagnóstico.
Também foi relatado um tipo de MRS monossintomático que apresenta apenas edema palpebral como achado oftalmológico 3). Outros achados oftalmológicos associados incluem neurite óptica retrobulbar, opacidade corneana, ceratoconjuntivite seca e blefarocalásia 3). Complicações sistêmicas como diverticulite e uveíte foram relatadas, sugerindo associação com doença de Crohn e sarcoidose.
QComo diferenciar paralisia do nervo facial de paralisia de Bell?
A
A paralisia de Bell não é acompanhada de edema orofacial. Já na MRS, o edema orofacial está presente em 80-100% dos casos. Se a paralisia facial recorrente for acompanhada de edema orofacial, deve-se suspeitar fortemente de MRS.
A etiologia é desconhecida. Acredita-se que os seguintes fatores estejam envolvidos, mas não foi encontrada uma associação forte que explique a causa da MRS.
Os fatores desencadeantes e de risco são classificados da seguinte forma.
Classificação
Fatores principais
Fatores genéticos
Herança autossômica dominante (9p11), mutação no gene FATP1/SLC27A1
Infecção e imunidade
EBV, VZV, CMV, HSV, COVID-19, resposta imune celular tipo Th1
Quanto aos fatores genéticos, foi relatada herança autossômica dominante com o gene causador localizado no braço curto do cromossomo 9 (9p11). A mutação no gene ATP1 (FATP1) foi relatada em uma família chinesa, e a mutação heterozigótica SLC27A1 (FATP1) também foi relatada, mas não confirmada 3). Em relatos de casos familiares de MRS, língua fissurada foi observada no irmão, filho, mãe e irmã do paciente 5).
Quanto às doenças infecciosas, infecções virais (EBV, VZV, CMV, HSV) podem ser desencadeantes. Um caso de recidiva após 4 anos desencadeado pela infecção por COVID-19 foi relatado, sugerindo que a ativação de mastócitos pode estar envolvida em comum na resposta inflamatória da COVID-19 e na patogênese da MRS2).
Imunologicamente, acredita-se que a resposta imune celular do tipo Th1 esteja envolvida 1). Os alérgenos incluem alimentos que contêm glúten, chocolate, canela, aditivos alimentares (glutamato monossódico), pasta de dente, metais (ouro, mercúrio, cobalto, zinco) e amálgama dentário 1).
A sobreposição com doença de Crohn e sarcoidose tem sido discutida como doenças relacionadas. No diagnóstico de sarcoidose, a MRS é listada como uma das “outras doenças granulomatosas cutâneas” que devem ser excluídas, exigindo a diferenciação entre ambas.
QSe houver alguém na família com os mesmos sintomas, existe uma relação genética?
A
Na SMR, sugere-se herança autossômica dominante, e foram relatados casos intrafamiliares de língua fissurada e paralisia do nervo facial 5). No entanto, a diversidade clínica e genética da doença é grande, e a causa genética ainda não foi determinada 3).
O diagnóstico clínico baseado na história e no exame físico é fundamental. O diagnóstico é considerado suficiente se pelo menos dois dos três sintomas principais estiverem presentes, ou se a biópsia confirmar queilite granulomatosa 5). O diagnóstico definitivo é raro, sendo necessário acompanhamento a longo prazo.
Os seguintes exames são realizados para diagnóstico diferencial.
Cálcio sérico e atividade da ECA: Normais para excluir sarcoidose1)
Radiografia de tórax e TCAR: Verificar presença de linfadenopatia hilar (excluir sarcoidose)
TC e RM: Confirmar espessamento de tecidos moles. A RM também é usada para avaliar o nervo facial3)
Exame eletrofisiológico nervoso: Estudo de condução do nervo motor facial para confirmar redução da amplitude do potencial de ação5)
Se houver suspeita de sarcoidose com base em achados oculares, realize exames de sangue (ECA sérica, receptor solúvel de IL-2 sérico) e radiografia de tórax, e encaminhe ao pneumologista ou dermatologista conforme necessário.
Outros diagnósticos diferenciais incluem hipotireoidismo, síndrome da veia cava superior, linfangioma, erisipela e linfoma. O diagnóstico pode exigir colaboração multidisciplinar entre oftalmologia, dermatologia, imunologia e gastroenterologia.
Não há tratamento curativo e a recorrência é inevitável. Remissão espontânea foi relatada, mas se não tratado, os episódios de sintomas podem se tornar mais frequentes e prolongados. O tratamento padrão não está estabelecido, e a seguinte abordagem gradual foi relatada.
Injeção de esteroide intralesional: Triancinolona acetonida 10-40 mg/mL. Mostrou eficácia equivalente à administração sistêmica1)3).
Esteroides orais: Prednisona (1 mg/kg/dia, por 10 dias, depois redução gradual). Alta taxa de remissão, mas recidiva5).
AINEs e anti-histamínicos: Usados como tratamento inicial.
Segunda linha em diante
Antibióticos: Minociclina 100 mg/dia (eficaz em combinação com esteroides)1)2). Terapia pulsátil com azitromicina (500 mg/dia × 3 dias/semana) também foi tentada.
Imunossupressores e agentes biológicos: Clofazimina, metotrexato, dapsona, sulfassalazina. Infliximabe (com base na semelhança com a doença de Crohn) 1).
Pomada de tacrolimo 0,03%: Eficaz em casos de edema palpebral refratários a esteroides3).
Medicamentos antivirais (aciclovir 400 mg a cada 4 horas) podem ser usados em combinação com corticosteroides para paralisia do nervo facial5).
Descompressão do nervo facial: Indicada para paralisia facial persistente ou recorrente refratária ao tratamento conservador. Considerada em grau IV ou superior na escala de House-Brackmann. Há relatos de melhora do grau VI para o grau II (8 meses após a cirurgia), e também sugere-se efeito preventivo contra recorrência futura5). A descompressão tardia após 90 dias também é considerada eficaz5).
Queiloplastia (cheiloplasty): Realizada para inchaço labial refratário.
Irradiação com laser de hélio-neônio: Há relatos de benefício em pacientes com duração da doença inferior a 4 anos.
Radioterapia: Eficácia consistente não foi demonstrada.
QA cirurgia é eficaz se a terapia medicamentosa for insuficiente?
A
A cirurgia de descompressão do nervo facial é uma opção eficaz para paralisia facial resistente ao tratamento conservador. Foram relatados casos de melhora dramática de paralisia grave (Grau VI de House-Brackmann) para Grau II 5), e também é sugerida para prevenir recorrência futura. Ensaios controlados com tratamento conservador não foram realizados, mas a cirurgia deve ser considerada para paralisia persistente de Grau IV ou superior.
6. Fisiopatologia e Mecanismo Detalhado de Ocorrência
A fisiopatologia básica da MRS é uma inflamação granulomatosa crônica composta principalmente por granulomas epitelioides não caseosos. Histologicamente, é acompanhada por células gigantes multinucleadas do tipo Langerhans, infiltrado inflamatório mononuclear perivascular, linfedema e fibrose.
Do ponto de vista imunológico, foi demonstrado o envolvimento da resposta imune celular do tipo Th1 1). Acredita-se que a ativação de mastócitos estimule a liberação de citocinas (IL-1, IL-6), causando inflamação granulomatosa local e edema 2).
O mecanismo da paralisia do nervo facial é a compressão do tecido nervoso por infiltração granulomatosa e edema. A cada recorrência, o nervo é progressivamente danificado, prolongando a duração da paralisia 5).
A base genética sugere mutações no gene FATP1 (SLC27A1) na região 9p11, mas a grande diversidade clínica e genética da doença impede conclusões definitivas 3).
Discute-se a possibilidade de que MRS, queilite granulomatosa, sarcoidose e doença de Crohn constituam um espectro contínuo 1). Todas essas doenças são caracterizadas por granulomas não caseosos decorrentes de resposta imune do tipo Th1 e apresentam semelhanças clínicas e histopatológicas.
7. Pesquisas mais recentes e perspectivas futuras (relatórios em fase de pesquisa)
Talidere et al. (2021) relataram uma mulher de 51 anos que apresentou recidiva da SMR após 4 anos devido à infecção por COVID-192). Ela apresentou a tríade clássica de edema labial, paralisia do nervo facial direito e língua fissurada, e melhorou com terapia com hidroxicloroquina, azitromicina e esteroides. Discutiu-se a possibilidade de os mastócitos desempenharem um papel comum nas reações inflamatórias da COVID-19 e da SMR, e foi relatado pela primeira vez que a infecção por COVID-19 pode ser um gatilho para a recidiva da SMR.
Estacia et al. (2022) relataram melhora em uma mulher de 59 anos com MRS de sintoma único apresentando apenas edema palpebral direito, que não respondeu a cinco injeções intralesionais de triancinolona (ao longo de 1 ano) e prednisona oral, após aplicação de pomada de tacrolimo a 0,03% 3). Essa nova terapia tópica é promissora para edema palpebral refratário a esteroides.
Resultados da Descompressão do Nervo Facial e MRS Familiar
Alencar et al. (2023) relataram melhora acentuada para Grau II, 8 meses após descompressão do nervo facial para o terceiro ataque (paralisia Grau VI à esquerda) de SMR familiar (mulher de 38 anos)5). O irmão, filho, mãe e irmã da paciente apresentavam língua fissurada, e o irmão tinha histórico de paralisia facial, sugerindo padrão de herança autossômica dominante. Este caso não melhorou com tratamento conservador (prednisona 60 mg/dia, aciclovir 400 mg), indicando eficácia da descompressão tardia e efeito preventivo de recorrência futura.
A mutação heterozigótica do SLC27A1 (FATP1) foi relatada como um gene candidato para a causa da MRS, mas devido à grande diversidade clínica e genética, é difícil explicar toda a doença com uma única mutação genética 3). Espera-se que o avanço da pesquisa genética elucide a patogênese da MRS e identifique alvos terapêuticos.
Tummidi S, Nagendran P, Anthony ML, Ramani RJ, Shankaralingappa A, Gopinath H. Granulomatous cheilitis of Miescher: a rare entity. BMC women’s health. 2023;23(1):118. doi:10.1186/s12905-023-02280-9. PMID:36944970; PMCID:PMC10031989.
Taşlıdere B, Mehmetaj L, Özcan AB, Gülen B, Taşlıdere N. Melkersson-Rosenthal Syndrome Induced by COVID-19. The American journal of emergency medicine. 2021;41:262.e5-262.e7. doi:10.1016/j.ajem.2020.08.018. PMID:32829989; PMCID:PMC7428670.
Estacia CT, Gameiro Filho AR, da Silveira IBE, Gameiro RR, Barba ALSD. Melkersson-Rosenthal syndrome: a rare variant of the monosymptomatic form. GMS ophthalmology cases. 2022;12:Doc04. doi:10.3205/oc000191. PMID:35291587; PMCID:PMC8900159.
Aukerman E, List M, Avashia-Khemka N. Melkersson-Rosenthal syndrome of the vulva. JAAD case reports. 2022;27:35-37. doi:10.1016/j.jdcr.2022.07.006. PMID:35996444; PMCID:PMC9391513.
Alencar TN, Botelho MM, Carasek N, Bahmad F Jr. Surgical Treatment Outcome for Familial Melkersson-Rosenthal Syndrome. The American journal of case reports. 2023;24:e938670. doi:10.12659/AJCR.938670. PMID:36755481; PMCID:PMC9923774.
Copie o texto do artigo e cole no assistente de IA de sua preferência.
Artigo copiado para a área de transferência
Abra um assistente de IA abaixo e cole o texto copiado na conversa.