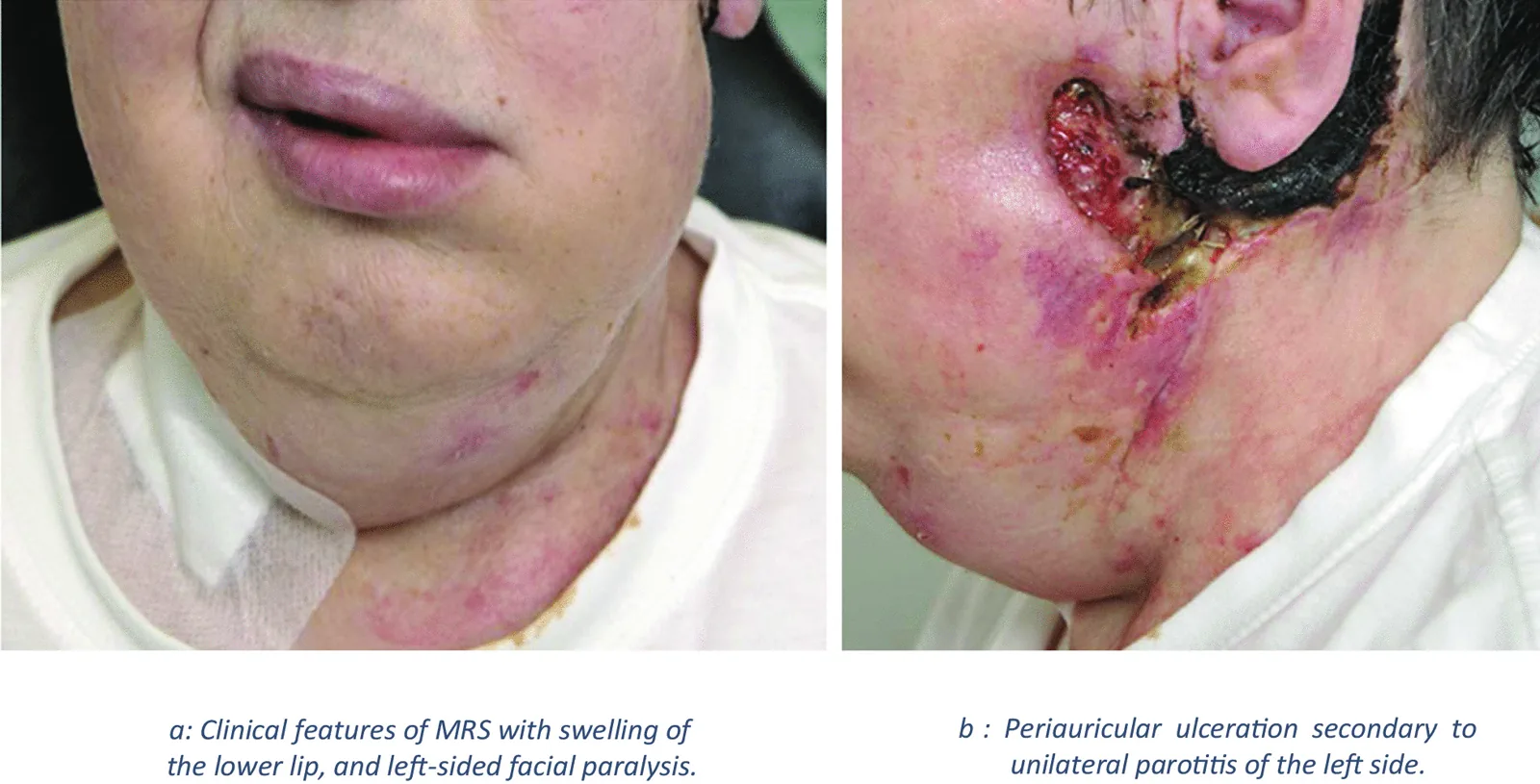
La sindrome di Melkersson-Rosenthal (MRS) è una malattia neurocutanea granulomatosa cronica non caseosa. Si presenta con edema orale e facciale, e la biopsia mostra infiammazione granulomatosa. Appartiene al gruppo di malattie chiamate «granulomatosi orofacciale».
Le descrizioni storiche di questa malattia sono le seguenti. Nel 1928 Ernst Melkersson riportò per la prima volta in dettaglio l’esistenza di paralisi facciale ricorrente ed edema. Nel 1931 Curt Rosenthal aggiunse la descrizione della lingua plicata (lingua plicata), e nel 1949 Lüscher usò per la prima volta il nome «sindrome di Melkersson-Rosenthal»3).
Dal punto di vista epidemiologico, si stima che si verifichi nello 0,08% della popolazione generale2). Si osserva più frequentemente tra i 20 e i 30 anni, con un’età media di insorgenza di 39 anni (range: 8-79 anni). Si riscontra una predominanza femminile (circa 3 volte).
La triade classica comprende i seguenti tre elementi.
Edema orofacciale ricorrente: in particolare gonfiore delle labbra (cheilite granulomatosa)
Paralisi facciale periferica ricorrente
Lingua plicata (lingua plicata)
Tutti e tre i sintomi sono presenti solo nell’8-25% dei casi. La maggior parte dei pazienti presenta solo 1-2 sintomi e la forma monosintomatica è talvolta descritta come cheilite granulomatosa (cheilite di Miescher)1).
Alcuni ricercatori suggeriscono che questa condizione possa essere un sintomo di sarcoidosi o morbo di Crohn, e la questione se si tratti di una malattia indipendente rimane irrisolta.
QÈ possibile diagnosticare solo se tutti e tre i sintomi sono presenti?
A
La triade classica è completa solo nell’8-25% dei casi. La diagnosi è possibile anche se 1-2 sintomi sono confermati dall’anamnesi e dall’esame obiettivo, o se una biopsia mostra cheilite granulomatosa. Una diagnosi definitiva è rara ed è necessario un follow-up a lungo termine.
J Casper, S Mohammad-Khani, J J Schmidt et al. Melkersson–Rosenthal syndrome in the context of sarcoidosis: a case report . Journal of Medical Case Reports. 2021 Oct 4; 15:488. Figure 2. PMCID: PMC8489098. License: CC BY.
Figura: Immagine che mostra le caratteristiche della sindrome di Melkersson-Rosenthal
I tre elementi della triade differiscono notevolmente per frequenza e caratteristiche.
Edema orofacciale
Frequenza: la più alta, si verifica nell’80-100% dei casi.
Caratteristiche tipiche: unilaterale, indolore, non comprimibile. Predilige il labbro superiore.
Decorso: inizialmente transitorio e intermittente, ma può diventare persistente. Possibile fibrosi tissutale.
Diagnosi differenziale: simile all’angioedema, ma MRS è più persistente e non risponde agli antistaminici. Complemento normale.
Paralisi del nervo facciale
Frequenza: 47–60% dei casi (fino al 90%). Unilaterale o bilaterale.
Decorso: il primo episodio spesso si risolve, ma con le recidive la durata si prolunga e può diventare permanente. Tasso di recidiva 3–11%.
Relazione con l’edema: può manifestarsi alcuni anni prima o dopo l’edema orofacciale. Nel 13-50% dei soggetti affetti, l’edema e la paralisi faciale sono associati.
Epidemiologia: l’incidenza della paralisi faciale correlata a MRS è di 0,36 persone/anno per 100.000 abitanti5).
Lingua scrotale
Frequenza: 30-35% dei casi.
Caratteristiche: solchi profondi circa 2 mm e lunghi circa 15 mm. Può essere congenita.
Significato diagnostico: essendo presente anche nel 5% della popolazione normale, da sola non costituisce un criterio diagnostico.
Come reperto oftalmologico, è stato riportato anche un MRS monosintomatico che presenta solo edema palpebrale 3). Altri reperti oftalmologici associati includono neurite ottica retrobulbare, opacità corneale, cheratocongiuntivite secca e blefarocalasi 3). Come complicanze sistemiche sono state riportate diverticolite e uveite, suggerendo un’associazione con il morbo di Crohn e la sarcoidosi.
QCome si distingue la paralisi facciale dalla paralisi di Bell?
A
La paralisi di Bell non è accompagnata da edema orofacciale. Nel MRS, invece, l’edema orofacciale è presente nell’80-100% dei casi. In caso di paralisi facciale ricorrente accompagnata da edema orofacciale, si deve sospettare attivamente un MRS.
L’eziologia è sconosciuta. Si ritiene che i seguenti fattori siano coinvolti, ma non è stata trovata una forte associazione che spieghi la causa del MRS.
I fattori scatenanti e di rischio sono classificati come segue.
Classificazione
Fattori principali
Fattori genetici
Ereditarietà autosomica dominante (9p11), mutazione del gene FATP1/SLC27A1
Infezione e immunità
EBV, VZV, CMV, HSV, COVID-19, risposta immunitaria cellulare di tipo Th1
Stress, cambiamenti ormonali, ipersensibilità ai UV-B, atopia
Per quanto riguarda i fattori genetici, è stata riportata una trasmissione autosomica dominante con un gene causale sul braccio corto del cromosoma 9 (9p11). Una mutazione del gene ATP1 (FATP1) è stata descritta in una famiglia cinese, e anche una mutazione eterozigote SLC27A1 (FATP1) è stata riportata, ma senza conferma definitiva3). Nei casi familiari di MRS, è stata osservata lingua scrotale nel fratello, nel figlio, nella madre e nella sorella del paziente5).
Per quanto riguarda le infezioni, le infezioni virali (EBV, VZV, CMV, HSV) possono essere fattori scatenanti. È stato riportato un caso di recidiva dopo 4 anni a seguito di un’infezione da COVID-19, suggerendo che l’attivazione dei mastociti potrebbe essere coinvolta sia nella risposta infiammatoria del COVID-19 che nella patologia della MRS2).
Dal punto di vista immunologico, si ritiene che sia coinvolta una risposta immunitaria cellulare di tipo Th1 1). Gli allergeni includono alimenti contenenti glutine, cioccolato, cannella, additivi alimentari (glutammato monosodico), dentifricio, metalli (oro, mercurio, cobalto, zinco) e amalgama dentale 1).
Si discute di una sovrapposizione con il morbo di Crohn e la sarcoidosi come malattie correlate. Nella diagnosi di sarcoidosi, la MRS è elencata come una delle «altre malattie cutanee granulomatose» da escludere, e la differenziazione tra le due è necessaria.
QSe in famiglia ci sono gli stessi sintomi, c'è una correlazione genetica?
A
Nella SMR si ipotizza una trasmissione autosomica dominante e sono stati riportati casi di lingua scrotale o paralisi facciale all’interno delle famiglie 5). Tuttavia, la diversità clinica e genetica della malattia è ampia e la causa genetica non è stata ancora determinata 3).
La diagnosi clinica si basa sull’anamnesi e sull’esame obiettivo. La presenza di almeno due dei tre segni cardinali, o la conferma di cheilite granulomatosa tramite biopsia, è considerata sufficiente per la diagnosi 5). Una diagnosi definitiva è rara ed è necessario un follow-up a lungo termine.
Per escludere infezioni, si eseguono colorazione delle sinechie anteriori dell’iride periferica, colorazione di Ziehl-Neelsen, colorazione argentica di Grocott, ecc.1)4).
Per la diagnosi differenziale vengono eseguiti i seguenti esami.
Calcio sierico e attività dell’ACE : la normalità esclude la sarcoidosi1)
Radiografia del torace e HRCT : verifica della presenza di linfoadenopatia ilare (esclusione della sarcoidosi)
TC e RM : conferma dell’ispessimento dei tessuti molli. La RM viene utilizzata anche per la valutazione del nervo facciale3)
Esame elettrofisiologico nervoso : conferma della riduzione dell’ampiezza del potenziale d’azione mediante lo studio della conduzione del nervo facciale5)
Se si sospetta una sarcoidosi sulla base dei reperti oculari, devono essere eseguiti esami del sangue (ACE sierico, recettore solubile dell’IL-2) e una radiografia del torace, e il paziente deve essere indirizzato a uno pneumologo o dermatologo se necessario.
Altre diagnosi differenziali includono ipotiroidismo, sindrome della vena cava superiore, linfangioma, erisipela e linfoma. La diagnosi può richiedere una collaborazione multidisciplinare tra oftalmologia, dermatologia, immunologia e gastroenterologia.
Non esiste una terapia curativa e le recidive sono inevitabili. Sono state riportate remissioni spontanee, ma senza trattamento gli episodi sintomatici possono diventare più frequenti e prolungati. Non esiste una terapia standard stabilita; è stato riportato il seguente approccio graduale.
Iniezione intralesionale di steroidi : triamcinolone acetonide 10–40 mg/mL. Efficacia dimostrata equivalente alla somministrazione sistemica1)3).
Steroidi orali : prednisone (1 mg/kg/die, poi riduzione graduale dopo 10 giorni). Alto tasso di remissione ma recidive5).
FANS e antistaminici : utilizzati come terapia iniziale.
Seconda linea e successive
Antibiotici : minociclina 100 mg/die (efficace in combinazione con steroidi)1)2). È stata anche tentata la terapia pulsata con azitromicina (500 mg/die × 3 giorni/settimana).
Farmaci immunosoppressori e agenti biologici : clofazimina, metotrexato, dapsone, sulfasalazina. Infliximab (basato sulla somiglianza con il morbo di Crohn) 1).
Unguento di tacrolimus allo 0,03% : efficace nell’edema palpebrale refrattario agli steroidi3).
I farmaci antivirali (aciclovir 400 mg ogni 4 ore) possono essere usati in combinazione con i corticosteroidi per la paralisi del nervo facciale 5).
Decompressione del nervo facciale : indicata per la paralisi facciale persistente o ricorrente refrattaria al trattamento conservativo. Considerata per grado House-Brackmann IV o superiore. Sono stati riportati miglioramenti dal grado VI al grado II (8 mesi post-operatori), e si suggerisce un effetto preventivo contro future recidive 5). Anche la decompressione ritardata dopo 90 giorni è considerata efficace 5).
Cheiloplastica : eseguita per gonfiore labiale refrattario.
Irradiazione con laser elio-neon: ci sono segnalazioni di beneficio in pazienti con durata della malattia inferiore a 4 anni.
Radioterapia: non è stata dimostrata un’efficacia costante.
QLa chirurgia è efficace quando la terapia farmacologica è insufficiente?
A
Per la paralisi facciale resistente al trattamento conservativo, la decompressione del nervo facciale è un’opzione efficace. Sono stati riportati miglioramenti drammatici da paralisi grave (Grado VI di House-Brackmann) a Grado II5), ed è anche suggerito un effetto preventivo contro future recidive. Non sono stati condotti studi controllati rispetto al trattamento conservativo, ma in caso di paralisi persistente di Grado IV o superiore, si dovrebbe prendere in considerazione un intervento chirurgico.
6. Fisiopatologia e meccanismo dettagliato della malattia
La patologia di base della MRS è un’infiammazione granulomatosa cronica caratterizzata principalmente da granulomi epitelioidi non caseosi. Istologicamente, si accompagna a cellule giganti multinucleate di tipo Langhans, infiltrati infiammatori mononucleati perivascolari, linfedema e fibrosi.
Dal punto di vista immunologico, è stato dimostrato il coinvolgimento della risposta immunitaria cellulo-mediata di tipo Th1 1). L’attivazione dei mastociti favorisce il rilascio di citochine (IL-1, IL-6), ritenuto responsabile dell’induzione di infiammazione granulomatosa locale ed edema 2).
Il meccanismo della paralisi facciale è la compressione del tessuto nervoso dovuta a infiltrazione granulomatosa ed edema. Ogni recidiva danneggia progressivamente il nervo, prolungando la durata della paralisi 5).
La base genetica suggerisce mutazioni nella regione 9p11 e nel gene FATP1 (SLC27A1), ma la diversità clinica e genetica della malattia è ampia e non è stata raggiunta una conclusione definitiva 3).
Si discute che MRS, cheilite granulomatosa, sarcoidosi e morbo di Crohn possano costituire uno spettro continuo 1). Queste malattie sono tutte caratterizzate da granulomi non caseosi dovuti a una risposta immunitaria di tipo Th1 e presentano somiglianze cliniche e istopatologiche.
7. Ricerche recenti e prospettive future (rapporti di fase di ricerca)
Talidere et al. (2021) hanno riportato il caso di una donna di 51 anni con recidiva di sindrome di Melkersson-Rosenthal (MRS) dopo 4 anni, in seguito a infezione da COVID-192). Presentava la triade classica completa: edema labiale, paralisi del nervo facciale destro e lingua scrotale, e migliorava con terapia a base di idrossiclorochina, azitromicina e steroidi. È stato ipotizzato che i mastociti possano svolgere un ruolo comune nella risposta infiammatoria di COVID-19 e MRS, ed è stato riportato per la prima volta che l’infezione da COVID-19 può essere un fattore scatenante della recidiva di MRS.
Applicazione del tacrolimus topico all’edema palpebrale
Estacia et al. (2022) hanno riportato un miglioramento in una donna di 59 anni con edema palpebrale destro isolato (MRS monosintomatica), refrattaria a 5 iniezioni intralesionali di triamcinolone (in 1 anno) e a prednisone orale, dopo applicazione di unguento di tacrolimus 0,03%3). Questo è considerato una nuova terapia locale per l’edema palpebrale resistente agli steroidi.
Risultati della decompressione del nervo facciale e MRS familiare
Alencar et al. (2023) hanno riportato un marcato miglioramento (Grado II) dopo decompressione del nervo facciale durante il terzo attacco (paralisi di Grado VI a sinistra) in una donna di 38 anni con MRS familiare, a 8 mesi dall’intervento 5). Il fratello, il figlio, la madre e la sorella della paziente presentavano lingua scrotale, e il fratello aveva una storia di paralisi facciale, suggerendo un pattern di ereditarietà autosomica dominante. Questo caso, che non aveva risposto al trattamento conservativo (prednisone 60 mg/die, aciclovir 400 mg), suggerisce l’efficacia della decompressione tardiva e un possibile effetto preventivo contro future recidive.
Una mutazione eterozigote di SLC27A1 (FATP1) è stata riportata come gene candidato per la MRS, ma a causa dell’ampia diversità clinica e genetica, è difficile spiegare l’intera malattia con una singola mutazione genica 3). I progressi nella ricerca genetica promettono di chiarire la patogenesi della MRS e identificare bersagli terapeutici.
Tummidi S, Nagendran P, Anthony ML, Ramani RJ, Shankaralingappa A, Gopinath H. Granulomatous cheilitis of Miescher: a rare entity. BMC women’s health. 2023;23(1):118. doi:10.1186/s12905-023-02280-9. PMID:36944970; PMCID:PMC10031989.
Taşlıdere B, Mehmetaj L, Özcan AB, Gülen B, Taşlıdere N. Melkersson-Rosenthal Syndrome Induced by COVID-19. The American journal of emergency medicine. 2021;41:262.e5-262.e7. doi:10.1016/j.ajem.2020.08.018. PMID:32829989; PMCID:PMC7428670.
Estacia CT, Gameiro Filho AR, da Silveira IBE, Gameiro RR, Barba ALSD. Melkersson-Rosenthal syndrome: a rare variant of the monosymptomatic form. GMS ophthalmology cases. 2022;12:Doc04. doi:10.3205/oc000191. PMID:35291587; PMCID:PMC8900159.
Aukerman E, List M, Avashia-Khemka N. Melkersson-Rosenthal syndrome of the vulva. JAAD case reports. 2022;27:35-37. doi:10.1016/j.jdcr.2022.07.006. PMID:35996444; PMCID:PMC9391513.
Alencar TN, Botelho MM, Carasek N, Bahmad F Jr. Surgical Treatment Outcome for Familial Melkersson-Rosenthal Syndrome. The American journal of case reports. 2023;24:e938670. doi:10.12659/AJCR.938670. PMID:36755481; PMCID:PMC9923774.
Copia il testo dell'articolo e incollalo nell'assistente IA che preferisci.
Articolo copiato negli appunti
Apri un assistente IA qui sotto e incolla il testo copiato nella chat.