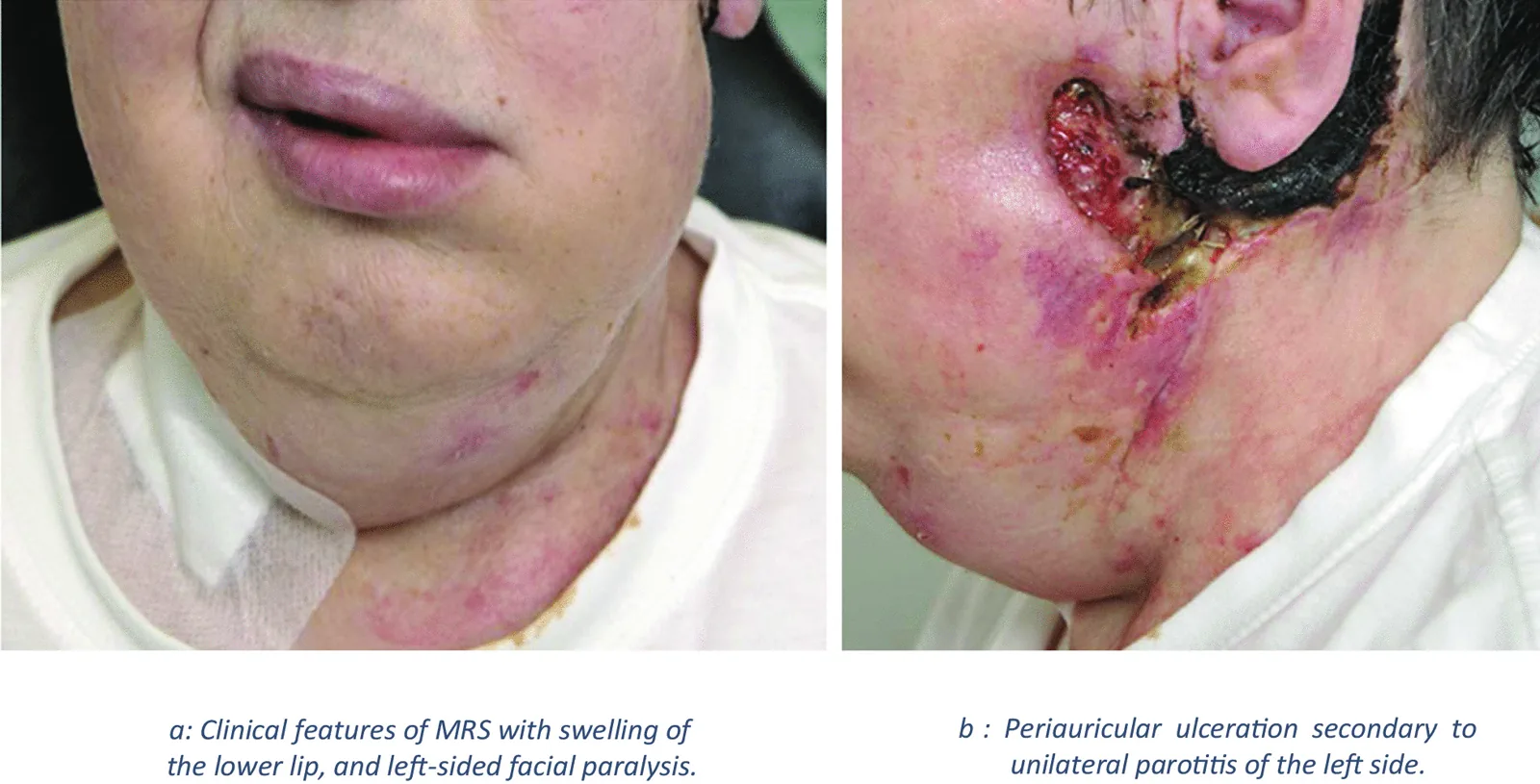
Le syndrome de Melkersson-Rosenthal (MRS) est une maladie neurocutanée granulomateuse chronique non caséeuse. Elle se manifeste par un œdème oral et facial, et une biopsie montre une inflammation granulomateuse. Elle appartient au groupe des « granulomatoses orofaciales ».
Les descriptions historiques de cette maladie sont les suivantes. En 1928, Ernst Melkersson a rapporté pour la première fois en détail l’existence d’une paralysie faciale récurrente et d’un œdème. En 1931, Curt Rosenthal a ajouté la description de la langue plicaturée (lingua plicata), et en 1949, Lüscher a utilisé pour la première fois le nom de « syndrome de Melkersson-Rosenthal »3).
Sur le plan épidémiologique, on estime qu’il survient chez 0,08 % de la population générale2). Il est le plus fréquent chez les personnes dans la vingtaine et la trentaine, avec un âge moyen d’apparition de 39 ans (extrêmes : 8 à 79 ans). Il y a une prédominance féminine (environ 3 fois).
La triade classique comprend les trois éléments suivants.
Œdème orofacial récurrent : en particulier gonflement des lèvres (chéilite granulomateuse)
Paralysie faciale périphérique récurrente
Langue plicaturée (lingua plicata)
Les trois symptômes ne sont présents que dans 8 à 25 % des cas. La plupart des patients ne présentent qu’un ou deux symptômes, et la forme monosymptomatique est parfois décrite comme une chéilite granulomateuse (chéilite de Miescher)1).
Certains chercheurs suggèrent que cette affection pourrait être une manifestation de la sarcoïdose ou de la maladie de Crohn, et la question de savoir s’il s’agit d’une entité distincte reste non résolue.
QEst-ce que le diagnostic ne peut être posé que si les trois symptômes sont tous présents ?
A
La triade classique n’est complète que dans 8 à 25 % des cas. Le diagnostic est possible même si un ou deux symptômes sont confirmés par l’anamnèse et l’examen clinique, ou si une biopsie révèle une chéilite granulomateuse. Un diagnostic définitif est rare et une surveillance à long terme est nécessaire.
J Casper, S Mohammad-Khani, J J Schmidt et al. Melkersson–Rosenthal syndrome in the context of sarcoidosis: a case report . Journal of Medical Case Reports. 2021 Oct 4; 15:488. Figure 2. PMCID: PMC8489098. License: CC BY.
Figure : Image montrant les caractéristiques du syndrome de Melkersson-Rosenthal
Les trois éléments de la triade diffèrent considérablement en fréquence et en caractéristiques.
Œdème orofacial
Fréquence : la plus élevée, survenant dans 80 à 100 % des cas.
Caractéristiques typiques : unilatéral, indolore, non dépressible. Prédominance à la lèvre supérieure.
Évolution : initialement transitoire et intermittente, mais peut devenir persistante par la suite. Peut évoluer vers une fibrose tissulaire.
Diagnostic différentiel : similaire à l’angio-œdème, mais le MRS est plus persistant et ne répond pas aux antihistaminiques. Le complément est normal.
Paralysie faciale
Fréquence : 47 à 60 % des cas (jusqu’à 90 %). Unilatérale ou bilatérale.
Évolution : la première atteinte récupère souvent, mais avec les récidives, la durée s’allonge et peut devenir permanente. Taux de récidive de 3 à 11 %.
Relation avec l’œdème : peut apparaître plusieurs années avant ou après l’œdème orofacial. Chez 13 à 50 % des personnes atteintes, l’œdème et la paralysie faciale sont associés.
Épidémiologie : l’incidence de la paralysie faciale liée au MRS est de 0,36 personne par an pour 100 000 habitants5).
Langue plicaturée
Fréquence : 30 à 35 % des cas.
Caractéristiques : sillons d’environ 2 mm de profondeur et 15 mm de longueur. Peut être congénital.
Signification diagnostique : étant également présente chez 5 % de la population normale, elle ne constitue pas à elle seule un critère diagnostique.
En tant que signe ophtalmologique, un MRS à symptôme unique ne présentant qu’un œdème palpébral a également été rapporté 3). Les autres signes ophtalmologiques associés comprennent la névrite optique rétrobulbaire, l’opacité cornéenne, la kératoconjonctivite sèche et la blépharochalasis 3). Des complications systémiques telles que la diverticulite et l’uvéite ont été rapportées, suggérant une association avec la maladie de Crohn et la sarcoïdose.
QComment distinguer la paralysie faciale de la paralysie de Bell ?
A
La paralysie de Bell n’est pas accompagnée d’œdème orofacial. En revanche, dans le MRS, un œdème orofacial est observé dans 80 à 100 % des cas. En cas de paralysie faciale récurrente accompagnée d’œdème orofacial, le MRS doit être activement suspecté.
L’étiologie est inconnue. Les facteurs suivants sont considérés comme impliqués, mais aucune association forte expliquant la cause du MRS n’a été trouvée.
Les facteurs déclenchants et de risque sont classés comme suit.
Classification
Facteurs principaux
Facteurs génétiques
Hérédité autosomique dominante (9p11), mutation du gène FATP1/SLC27A1
Infection et immunité
EBV, VZV, CMV, HSV, COVID-19, réponse immunitaire cellulaire de type Th1
Stress, changements hormonaux, hypersensibilité aux UV-B, atopie
En ce qui concerne les facteurs génétiques, une transmission autosomique dominante avec un gène causal sur le bras court du chromosome 9 (9p11) a été rapportée. Une mutation du gène ATP1 (FATP1) a été signalée dans une famille chinoise, et une mutation hétérozygote SLC27A1 (FATP1) a également été rapportée, mais sans confirmation définitive3). Dans les cas familiaux de MRS, une langue plicaturée a été observée chez le frère, le fils, la mère et la sœur du patient5).
En ce qui concerne les infections, les infections virales (EBV, VZV, CMV, HSV) peuvent être des déclencheurs. Un cas de récidive après 4 ans suite à une infection par le COVID-19 a été rapporté, suggérant que l’activation des mastocytes pourrait être impliquée à la fois dans la réponse inflammatoire du COVID-19 et dans la pathologie du MRS2).
Sur le plan immunologique, on pense qu’une réponse immunitaire cellulaire de type Th1 est impliquée 1). Les allergènes comprennent les aliments contenant du gluten, le chocolat, la cannelle, les additifs alimentaires (glutamate monosodique), le dentifrice, les métaux (or, mercure, cobalt, zinc) et l’amalgame dentaire 1).
La maladie de Crohn et la sarcoïdose sont discutées comme maladies associées. Dans le diagnostic de la sarcoïdose, la MRS est mentionnée comme l’une des « autres maladies cutanées granulomateuses » à exclure, nécessitant une différenciation entre les deux.
QSi un membre de la famille présente les mêmes symptômes, y a-t-il un lien génétique ?
A
Dans le SMR, une transmission autosomique dominante est suggérée, et des cas de langue plicaturée ou de paralysie faciale ont été rapportés dans les familles 5). Cependant, la diversité clinique et génétique de la maladie est grande, et la cause génétique n’a pas encore été déterminée 3).
Le diagnostic clinique repose sur les antécédents médicaux et l’examen physique. La présence d’au moins deux des trois signes cardinaux, ou la confirmation d’une chéilite granulomateuse par biopsie, est considérée comme suffisante pour le diagnostic 5). Un diagnostic définitif est rare, et une surveillance à long terme est nécessaire.
Pour exclure une infection, on réalise une coloration de l’iris périphérique, une coloration de Ziehl-Neelsen, une coloration argentique de Grocott, etc.1)4).
Les examens suivants sont réalisés pour le diagnostic différentiel.
Calcium sérique et activité de l’ACE : leur normalité permet d’exclure la sarcoïdose1)
Radiographie thoracique et HRCT : vérification de la présence d’une adénopathie hilaire (exclusion de la sarcoïdose)
CT et IRM : confirmation de l’épaississement des tissus mous. L’IRM est également utilisée pour évaluer le nerf facial3)
Examen électrophysiologique nerveux : confirmation de la diminution de l’amplitude du potentiel d’action par l’étude de la conduction du nerf facial5)
Si une sarcoïdose est suspectée sur la base des signes oculaires, des analyses sanguines (ACE sérique, récepteur soluble de l’IL-2) et une radiographie thoracique doivent être réalisées, et le patient doit être orienté vers un pneumologue ou un dermatologue si nécessaire.
Les autres diagnostics différentiels incluent l’hypothyroïdie, le syndrome de la veine cave supérieure, le lymphangiome, l’érysipèle et le lymphome. Le diagnostic peut nécessiter une collaboration pluridisciplinaire entre ophtalmologie, dermatologie, immunologie et gastro-entérologie.
Il n’existe pas de traitement curatif et les récidives sont inévitables. Des rémissions spontanées ont été rapportées, mais en l’absence de traitement, les épisodes symptomatiques peuvent devenir plus fréquents et plus longs. Aucun traitement standard n’est établi ; l’approche par étapes suivante a été rapportée.
Injection intralésionnelle de stéroïdes : acétonide de triamcinolone 10 à 40 mg/mL. Efficacité démontrée équivalente à l’administration systémique1)3).
Corticostéroïdes oraux : prednisone (1 mg/kg/jour, puis diminution progressive après 10 jours). Taux de rémission élevé mais récidives fréquentes5).
AINS et antihistaminiques : utilisés en traitement initial.
Deuxième intention et suivantes
Antibiotiques : minocycline 100 mg/jour (efficace en association avec les stéroïdes)1)2). Une thérapie pulsée à l’azithromycine (500 mg/jour × 3 jours/semaine) a également été essayée.
Immunosuppresseurs et biothérapies : clofazimine, méthotrexate, dapsone, sulfasalazine. Infliximab (basé sur la similitude avec la maladie de Crohn) 1).
Pommade de tacrolimus à 0,03 % : efficace dans les cas d’œdème palpébral réfractaires aux stéroïdes3).
Les antiviraux (acyclovir 400 mg toutes les 4 heures) peuvent être associés aux corticostéroïdes pour la paralysie faciale 5).
Décompression du nerf facial : indiquée pour la paralysie faciale persistante ou récurrente réfractaire au traitement conservateur. Envisagée pour un grade House-Brackmann IV ou supérieur. Des améliorations du grade VI au grade II ont été rapportées (8 mois postopératoires), avec un effet préventif potentiel contre les récidives futures 5). Une décompression tardive après 90 jours est également considérée comme efficace 5).
Chéiloplastie : réalisée en cas de gonflement labial réfractaire.
Irradiation au laser hélium-néon : des rapports indiquent qu’elle est bénéfique chez les patients dont la durée de la maladie est inférieure à 4 ans.
Radiothérapie : aucune efficacité constante n’a été démontrée.
QLa chirurgie est-elle efficace lorsque le traitement médicamenteux est insuffisant ?
A
Pour la paralysie faciale résistante au traitement conservateur, la décompression du nerf facial est une option efficace. Des améliorations spectaculaires, passant d’une paralysie sévère (Grade VI de House-Brackmann) au Grade II, ont été rapportées5), et un effet préventif contre les récidives futures est également suggéré. Aucun essai contrôlé comparant au traitement conservateur n’a été réalisé, mais une intervention chirurgicale doit être envisagée en cas de paralysie persistante de Grade IV ou plus.
6. Physiopathologie et mécanisme détaillé de la maladie
La pathologie fondamentale du MRS est une inflammation granulomateuse chronique dominée par des granulomes épithélioïdes non caséeux. Histologiquement, elle s’accompagne de cellules géantes multinucléées de type Langhans, d’infiltrats inflammatoires mononucléés périvasculaires, de lymphœdème et de fibrose.
Sur le plan immunologique, l’implication de la réponse immunitaire à médiation cellulaire de type Th1 a été démontrée 1). L’activation des mastocytes favorise la libération de cytokines (IL-1, IL-6), ce qui est considéré comme provoquant une inflammation granulomateuse locale et un œdème 2).
Le mécanisme de la paralysie faciale est la compression du tissu nerveux par une infiltration granulomateuse et un œdème. Chaque récidive endommage progressivement le nerf, ce qui prolonge la durée de la paralysie 5).
La base génétique suggère des mutations dans la région 9p11 et le gène FATP1 (SLC27A1), mais la diversité clinique et génétique de la maladie est grande et aucune conclusion définitive n’a été atteinte 3).
Il est discuté que le MRS, la chéilite granulomateuse, la sarcoïdose et la maladie de Crohn pourraient constituer un spectre continu 1). Ces maladies sont toutes caractérisées par des granulomes non caséeux dus à une réponse immunitaire de type Th1 et présentent des similitudes cliniques et histopathologiques.
7. Recherches récentes et perspectives futures (rapports de stade de recherche)
Talidere et al. (2021) ont rapporté le cas d’une femme de 51 ans dont le syndrome de Melkersson-Rosenthal (MRS) a récidivé après 4 ans suite à une infection par la COVID-192). Elle présentait la triade classique complète : œdème des lèvres, paralysie faciale droite et langue plicaturée, et s’est améliorée avec un traitement par hydroxychloroquine, azithromycine et stéroïdes. Les auteurs ont émis l’hypothèse que les mastocytes pourraient jouer un rôle commun dans la réaction inflammatoire de la COVID-19 et du MRS, et ont rapporté pour la première fois que l’infection par la COVID-19 pourrait être un facteur déclenchant de la récidive du MRS.
Application du tacrolimus topique à l’œdème palpébral
Estacia et al. (2022) ont rapporté une amélioration après application de pommade de tacrolimus à 0,03% chez une femme de 59 ans présentant un œdème palpébral droit isolé (MRS monosymptomatique), réfractaire à 5 injections intralésionnelles de triamcinolone (sur un an) et à la prednisone orale3). Ce traitement est considéré comme une nouvelle thérapie locale pour l’œdème palpébral résistant aux stéroïdes.
Résultats de la décompression du nerf facial et MRS familiale
Alencar et al. (2023) ont rapporté une amélioration marquée (Grade II) après décompression du nerf facial lors de la troisième crise (paralysie Grade VI à gauche) chez une femme de 38 ans atteinte de MRS familiale, huit mois après l’opération 5). Le frère, le fils, la mère et la sœur de la patiente présentaient une langue plicaturée, et le frère avait des antécédents de paralysie faciale, suggérant un mode de transmission autosomique dominant. Ce cas, qui n’avait pas répondu au traitement conservateur (prednisone 60 mg/jour, aciclovir 400 mg), suggère l’efficacité de la décompression tardive et un possible effet préventif contre les récidives futures.
Une mutation hétérozygote de SLC27A1 (FATP1) a été rapportée comme gène candidat responsable du MRS, mais en raison de la grande diversité clinique et génétique, il est difficile d’expliquer l’ensemble de la maladie par une seule mutation génétique 3). Les progrès de la recherche génétique devraient permettre de mieux comprendre la pathogénie du MRS et d’identifier des cibles thérapeutiques.
Tummidi S, Nagendran P, Anthony ML, Ramani RJ, Shankaralingappa A, Gopinath H. Granulomatous cheilitis of Miescher: a rare entity. BMC women’s health. 2023;23(1):118. doi:10.1186/s12905-023-02280-9. PMID:36944970; PMCID:PMC10031989.
Taşlıdere B, Mehmetaj L, Özcan AB, Gülen B, Taşlıdere N. Melkersson-Rosenthal Syndrome Induced by COVID-19. The American journal of emergency medicine. 2021;41:262.e5-262.e7. doi:10.1016/j.ajem.2020.08.018. PMID:32829989; PMCID:PMC7428670.
Estacia CT, Gameiro Filho AR, da Silveira IBE, Gameiro RR, Barba ALSD. Melkersson-Rosenthal syndrome: a rare variant of the monosymptomatic form. GMS ophthalmology cases. 2022;12:Doc04. doi:10.3205/oc000191. PMID:35291587; PMCID:PMC8900159.
Aukerman E, List M, Avashia-Khemka N. Melkersson-Rosenthal syndrome of the vulva. JAAD case reports. 2022;27:35-37. doi:10.1016/j.jdcr.2022.07.006. PMID:35996444; PMCID:PMC9391513.
Alencar TN, Botelho MM, Carasek N, Bahmad F Jr. Surgical Treatment Outcome for Familial Melkersson-Rosenthal Syndrome. The American journal of case reports. 2023;24:e938670. doi:10.12659/AJCR.938670. PMID:36755481; PMCID:PMC9923774.
Copiez le texte de l'article et collez-le dans l'assistant IA de votre choix.
Article copié dans le presse-papiers
Ouvrez un assistant IA ci-dessous et collez le texte copié dans la conversation.