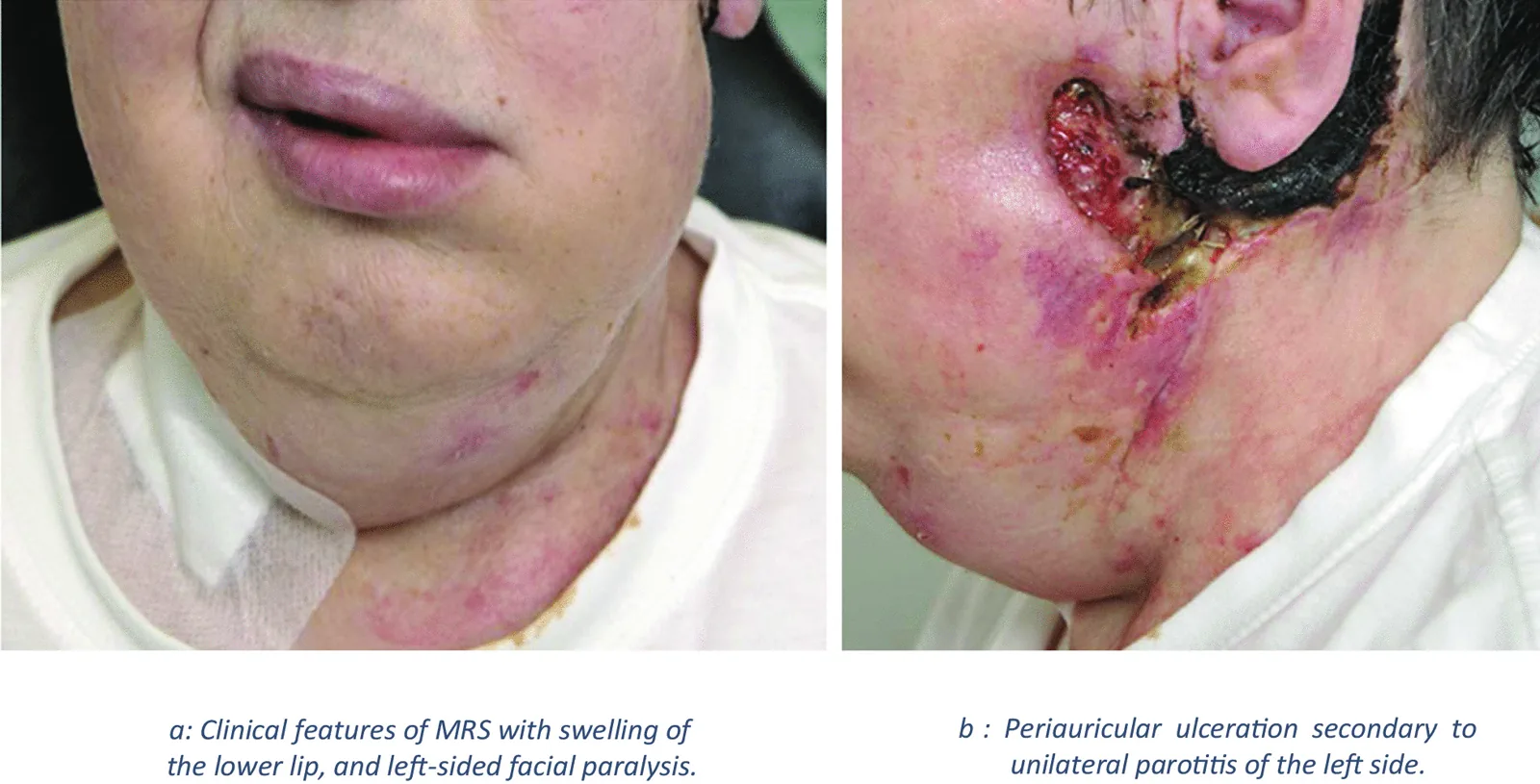
El síndrome de Melkersson-Rosenthal (MRS) es una enfermedad neurocutánea granulomatosa crónica no caseificante. Se presenta con edema oral y facial, y la biopsia muestra inflamación granulomatosa. Pertenece al grupo de enfermedades denominadas «granulomatosis orofacial».
La descripción histórica de esta enfermedad es la siguiente. En 1928, Ernst Melkersson informó por primera vez en detalle la existencia de parálisis facial recurrente y edema. En 1931, Curt Rosenthal agregó la lengua fisurada (lingua plicata), y en 1949, Lüscher utilizó por primera vez el nombre de «síndrome de Melkersson-Rosenthal» 3).
Epidemiológicamente, se estima que ocurre en el 0.08% de la población general 2). Se observa con mayor frecuencia en personas de 20 a 30 años, con una edad promedio de inicio de 39 años (rango: 8-79 años). Se considera que predomina en mujeres (aproximadamente 3 veces más).
La tríada clásica consta de los siguientes tres elementos:
Edema orofacial recurrente: especialmente hinchazón de los labios (queilitis granulomatosa).
Parálisis facial periférica recurrente
Lengua fisurada (lingua plicata)
Solo el 8-25% de los casos presentan los tres síntomas. La mayoría presenta solo uno o dos síntomas, y la forma monosintomática puede describirse como queilitis granulomatosa (queilitis de Miescher)1).
Algunos investigadores sugieren que esta enfermedad podría ser un síntoma de sarcoidosis o enfermedad de Crohn, y sigue sin resolverse si es una entidad independiente.
Q¿No se puede diagnosticar si no se presentan los tres síntomas?
A
La tríada clásica completa solo se observa en el 8-25% de los casos. El diagnóstico es posible si se confirman 1 o 2 síntomas mediante la historia clínica y el examen físico, o si se detecta queilitis granulomatosa en la biopsia. El diagnóstico definitivo es raro y requiere un seguimiento a largo plazo.
J Casper, S Mohammad-Khani, J J Schmidt et al. Melkersson–Rosenthal syndrome in the context of sarcoidosis: a case report . Journal of Medical Case Reports. 2021 Oct 4; 15:488. Figure 2. PMCID: PMC8489098. License: CC BY.
Los elementos de la tríada difieren considerablemente en frecuencia de presentación y características.
Edema orofacial
Frecuencia de aparición: la más alta, del 80 al 100% de los casos.
Características típicas: unilateral, indoloro, no deja fóvea. Predilección por el labio superior.
Evolución: inicialmente transitorio e intermitente, pero puede volverse persistente más tarde. Puede llevar a fibrosis del tejido.
Diagnóstico diferencial: Similar al angioedema, pero el MRS es más persistente y no responde a los antihistamínicos. El complemento sérico es normal.
Parálisis del nervio facial
Frecuencia de aparición: 47–60% de los casos (hasta 90%). Unilateral o bilateral.
Evolución: La primera vez suele recuperarse, pero con las recurrencias la duración se prolonga y puede volverse permanente. La tasa de recurrencia es del 3–11%.
Relación con el edema: puede aparecer varios años antes o después del edema orofacial. En el 13-50% de los afectados, el edema y la parálisis facial se presentan juntos.
Epidemiología: la incidencia de parálisis facial asociada a MRS es de 0.36 personas por cada 100,000 habitantes al año5).
Lengua fisurada
Frecuencia de aparición: 30-35% de los casos.
Características: surco de aproximadamente 2 mm de profundidad y 15 mm de longitud. Puede ser congénito en algunos casos.
Significado diagnóstico: se observa hasta en el 5% de la población normal, por lo que por sí solo no constituye un criterio diagnóstico.
Como hallazgo oftalmológico, también se ha reportado un MRS de tipo monosintomático que solo presenta edema palpebral3). Otros hallazgos oftalmológicos asociados incluyen neuritis retrobulbar, opacidad corneal, queratoconjuntivitis seca y blefarocalasia3). Como complicaciones sistémicas se han reportado diverticulitis y uveítis, y se sugiere una asociación con la enfermedad de Crohn y la sarcoidosis.
Q¿Cómo se diferencia la parálisis del nervio facial de la parálisis de Bell?
A
La parálisis de Bell no se acompaña de edema orofacial. En cambio, en el SMR se observa edema orofacial en el 80-100% de los casos. Si la parálisis facial recurrente se acompaña de edema orofacial, se debe sospechar fuertemente de SMR.
Se desconoce la etiología. Se cree que los siguientes factores están involucrados, pero no se ha encontrado una asociación sólida que explique la causa del SMR.
Los factores desencadenantes y de riesgo se clasifican de la siguiente manera.
Clasificación
Factores principales
Factores genéticos
Herencia autosómica dominante (9p11), mutación del gen FATP1/SLC27A1
Infección e inmunidad
EBV, VZV, CMV, HSV, COVID-19, respuesta inmune celular tipo Th1
Estrés, cambios hormonales, hipersensibilidad a UV-B, atopia
En cuanto a los factores genéticos, se ha reportado herencia autosómica dominante con el gen causal en el brazo corto del cromosoma 9 (9p11). Se ha informado de una mutación en el gen ATP1 (FATP1) en una familia china, y también se ha reportado una mutación heterocigota en SLC27A1 (FATP1), aunque no se ha confirmado definitivamente 3). En casos reportados de MRS familiar, se observó lengua fisurada en el hermano, hijo, madre y hermana del paciente 5).
En cuanto a las infecciones, las infecciones virales (EBV, VZV, CMV, HSV) pueden ser desencadenantes. Se ha reportado un caso de recurrencia después de 4 años desencadenado por la infección por COVID-19, lo que sugiere que la activación de los mastocitos podría estar involucrada tanto en la respuesta inflamatoria del COVID-19 como en la patología del MRS 2).
Inmunológicamente, se considera la participación de la respuesta inmune celular tipo Th11). Como alérgenos se incluyen alimentos que contienen gluten, chocolate, canela, aditivos alimentarios (glutamato monosódico), pasta de dientes, metales (oro, mercurio, cobalto, zinc) y amalgamas dentales1).
Se discute la superposición con enfermedades relacionadas como la enfermedad de Crohn y la sarcoidosis. En el diagnóstico de sarcoidosis, la MRS se menciona como una de las «otras enfermedades granulomatosas cutáneas» que deben excluirse, por lo que se requiere el diagnóstico diferencial entre ambas.
QSi hay familiares con los mismos síntomas, ¿existe una relación genética?
A
En la MRS se ha sugerido una herencia autosómica dominante, y se han reportado casos con lengua fisurada o parálisis del nervio facial en familias5). Sin embargo, la diversidad clínica y genética de la enfermedad es amplia, y no se ha llegado a una determinación genética definitiva3).
El diagnóstico clínico se basa en la historia clínica y el examen físico. Se considera suficiente para el diagnóstico si se presentan dos o más de los tres signos cardinales, o si una biopsia confirma queilitis granulomatosa5). El diagnóstico definitivo es raro y se requiere un seguimiento a largo plazo.
Para el diagnóstico diferencial se realizan las siguientes pruebas.
Calcio sérico y actividad de la ECA: si son normales, se descarta sarcoidosis1)
Radiografía de tórax y HRCT: para verificar la presencia de adenopatía hiliar (descartar sarcoidosis)
TC y RM: para confirmar el engrosamiento de los tejidos blandos. La RM también se utiliza para evaluar el nervio facial3)
Estudios neurofisiológicos: en la conducción nerviosa motora del nervio facial se confirma una disminución de la amplitud del potencial de acción5)
Cuando se sospecha sarcoidosis por hallazgos oculares, se deben realizar análisis de sangre (ACE sérica, receptor soluble de IL-2 en suero) y radiografía de tórax, y derivar al paciente a neumología o dermatología según sea necesario.
Otras enfermedades diferenciales incluyen hipotiroidismo, síndrome de vena cava superior, linfangioma, erisipela y linfoma. El diagnóstico puede requerir colaboración multidisciplinaria entre oftalmología, dermatología, inmunología y gastroenterología.
No existe un tratamiento curativo y las recurrencias son inevitables. Se han reportado remisiones espontáneas, pero si no se trata, los episodios sintomáticos pueden volverse más frecuentes y prolongados. No se ha establecido un tratamiento estándar, y se ha descrito el siguiente enfoque escalonado.
Inyección intralesional de esteroides: triamcinolona acetonida 10–40 mg/mL. Se ha demostrado una eficacia similar a la administración sistémica1)3).
Esteroides orales: prednisona (1 mg/kg/día durante 10 días, luego reducción gradual). La tasa de remisión es alta, pero hay recurrencias5).
AINEs y antihistamínicos: se utilizan como tratamiento inicial.
Segunda línea y posteriores
Antibióticos: minociclina 100 mg/día (eficaz en combinación con esteroides)1)2). También se ha probado la terapia con pulsos de azitromicina (500 mg/día × 3 días/semana).
Inmunosupresores y agentes biológicos: clofazimina, metotrexato, dapsona, sulfasalazina. Infliximab (basado en la similitud con la enfermedad de Crohn)1).
Ungüento de tacrolimus al 0.03%: eficaz en casos de edema palpebral resistentes a esteroides3).
Los antivirales (aciclovir 400 mg cada 4 horas) pueden combinarse con esteroides para la parálisis del nervio facial5).
Descompresión del nervio facial: indicada para parálisis facial persistente o recurrente que no responde al tratamiento conservador. Se considera en grados House-Brackmann IV o superiores. Se han reportado mejorías de grado VI a II (8 meses postoperatorios) y también sugiere un efecto preventivo de recurrencias futuras5). La descompresión tardía después de 90 días también se considera efectiva5).
Queiloplastia: se realiza para la hinchazón labial refractaria.
Irradiación con láser de helio-neón: se ha informado que es beneficiosa en pacientes con una duración de la enfermedad de menos de 4 años.
Radioterapia: no se ha demostrado una eficacia consistente.
QSi el tratamiento farmacológico es insuficiente, ¿es efectiva la cirugía?
A
Para la parálisis facial resistente al tratamiento conservador, la descompresión del nervio facial es una opción efectiva. Se han reportado casos de mejora dramática desde parálisis severa (House-Brackmann Grado VI) hasta Grado II5), y también se sugiere un efecto preventivo de recurrencias futuras. Aunque no se han realizado ensayos controlados comparativos con el tratamiento conservador, se considera que se debe considerar la cirugía en parálisis persistentes de Grado IV o superior.
6. Fisiopatología y mecanismo detallado de la enfermedad
La patología básica del MRS es una inflamación granulomatosa crónica con granulomas de células epitelioides no caseificantes como componente principal. Histológicamente, se acompaña de células gigantes multinucleadas tipo Langhans, infiltrado inflamatorio mononuclear perivascular, linfedema y fibrosis.
En cuanto al mecanismo inmunológico, se ha demostrado la participación de la respuesta inmune celular tipo Th1 1). Se cree que la activación de los mastocitos promueve la liberación de citoquinas (IL-1, IL-6), lo que induce inflamación granulomatosa local y edema 2).
El mecanismo de la parálisis del nervio facial es la compresión del tejido nervioso debido a la infiltración granulomatosa y el edema. Con cada recurrencia, el nervio se daña progresivamente, por lo que la duración de la parálisis se prolonga 5).
En cuanto a la base genética, se ha sugerido la región 9p11 y la mutación del gen FATP1 (SLC27A1), pero debido a la gran diversidad clínica y genética de la enfermedad, no se ha llegado a una conclusión definitiva 3).
Se discute la posibilidad de que el síndrome de Melkersson-Rosenthal, la queilitis granulomatosa, la sarcoidosis y la enfermedad de Crohn constituyan un espectro continuo 1). Todas estas enfermedades se caracterizan por granulomas no caseificantes debidos a una respuesta inmune tipo Th1 y presentan similitudes clínicas e histopatológicas.
7. Investigación actual y perspectivas futuras (informes en fase de investigación)
Talidere et al. (2021) reportaron el caso de una mujer de 51 años que presentó una recurrencia de MRS después de 4 años, desencadenada por una infección por COVID-192). Presentó la tríada clásica completa de edema labial, parálisis del nervio facial derecho y lengua fisurada, y mejoró con hidroxicloroquina, azitromicina y terapia con esteroides. Se discutió la posibilidad de que los mastocitos desempeñen un papel común en la reacción inflamatoria de COVID-19 y MRS, y se informó por primera vez que la infección por COVID-19 puede ser un desencadenante de la recurrencia de MRS.
Aplicación de tacrolimus tópico para el edema palpebral
Estacia et al. (2022) reportaron la mejoría de un caso de MRS monosintomático (mujer de 59 años) que solo presentaba edema palpebral derecho, refractario a 5 inyecciones intralesionales de triamcinolona (durante 1 año) y a prednisona oral, tras la aplicación de ungüento de tacrolimus al 0.03%3). Se destaca como una nueva terapia tópica para el edema palpebral refractario a esteroides.
Resultados de la descompresión del nervio facial y MRS familiar
Alencar et al. (2023) reportaron una mejoría notable a Grado II a los 8 meses postoperatorios tras realizar una descompresión del nervio facial en un tercer episodio (parálisis Grado VI del lado izquierdo) de una paciente de 38 años con MRS familiar 5). El hermano, hijo, madre y hermana de la paciente presentaban lengua fisurada, y el hermano tenía antecedentes de parálisis facial, mostrando un patrón de herencia autosómica dominante. Se trataba de un caso que no había mejorado con tratamiento conservador (prednisona 60 mg/día, aciclovir 400 mg), lo que sugiere la eficacia de la descompresión tardía y su potencial para prevenir futuras recurrencias.
Se ha reportado una mutación heterocigota en SLC27A1 (FATP1) como posible gen causante de MRS, pero existe una gran diversidad clínica y genética, por lo que es difícil explicar toda la enfermedad con una sola mutación génica 3). Se espera que el avance en la investigación genética permita dilucidar la patogénesis de la MRS e identificar dianas terapéuticas.
Tummidi S, Nagendran P, Anthony ML, Ramani RJ, Shankaralingappa A, Gopinath H. Granulomatous cheilitis of Miescher: a rare entity. BMC women’s health. 2023;23(1):118. doi:10.1186/s12905-023-02280-9. PMID:36944970; PMCID:PMC10031989.
Taşlıdere B, Mehmetaj L, Özcan AB, Gülen B, Taşlıdere N. Melkersson-Rosenthal Syndrome Induced by COVID-19. The American journal of emergency medicine. 2021;41:262.e5-262.e7. doi:10.1016/j.ajem.2020.08.018. PMID:32829989; PMCID:PMC7428670.
Estacia CT, Gameiro Filho AR, da Silveira IBE, Gameiro RR, Barba ALSD. Melkersson-Rosenthal syndrome: a rare variant of the monosymptomatic form. GMS ophthalmology cases. 2022;12:Doc04. doi:10.3205/oc000191. PMID:35291587; PMCID:PMC8900159.
Aukerman E, List M, Avashia-Khemka N. Melkersson-Rosenthal syndrome of the vulva. JAAD case reports. 2022;27:35-37. doi:10.1016/j.jdcr.2022.07.006. PMID:35996444; PMCID:PMC9391513.
Alencar TN, Botelho MM, Carasek N, Bahmad F Jr. Surgical Treatment Outcome for Familial Melkersson-Rosenthal Syndrome. The American journal of case reports. 2023;24:e938670. doi:10.12659/AJCR.938670. PMID:36755481; PMCID:PMC9923774.
Copia el texto del artículo y pégalo en el asistente de IA que prefieras.
Artículo copiado al portapapeles
Abre un asistente de IA abajo y pega el texto copiado en el chat.