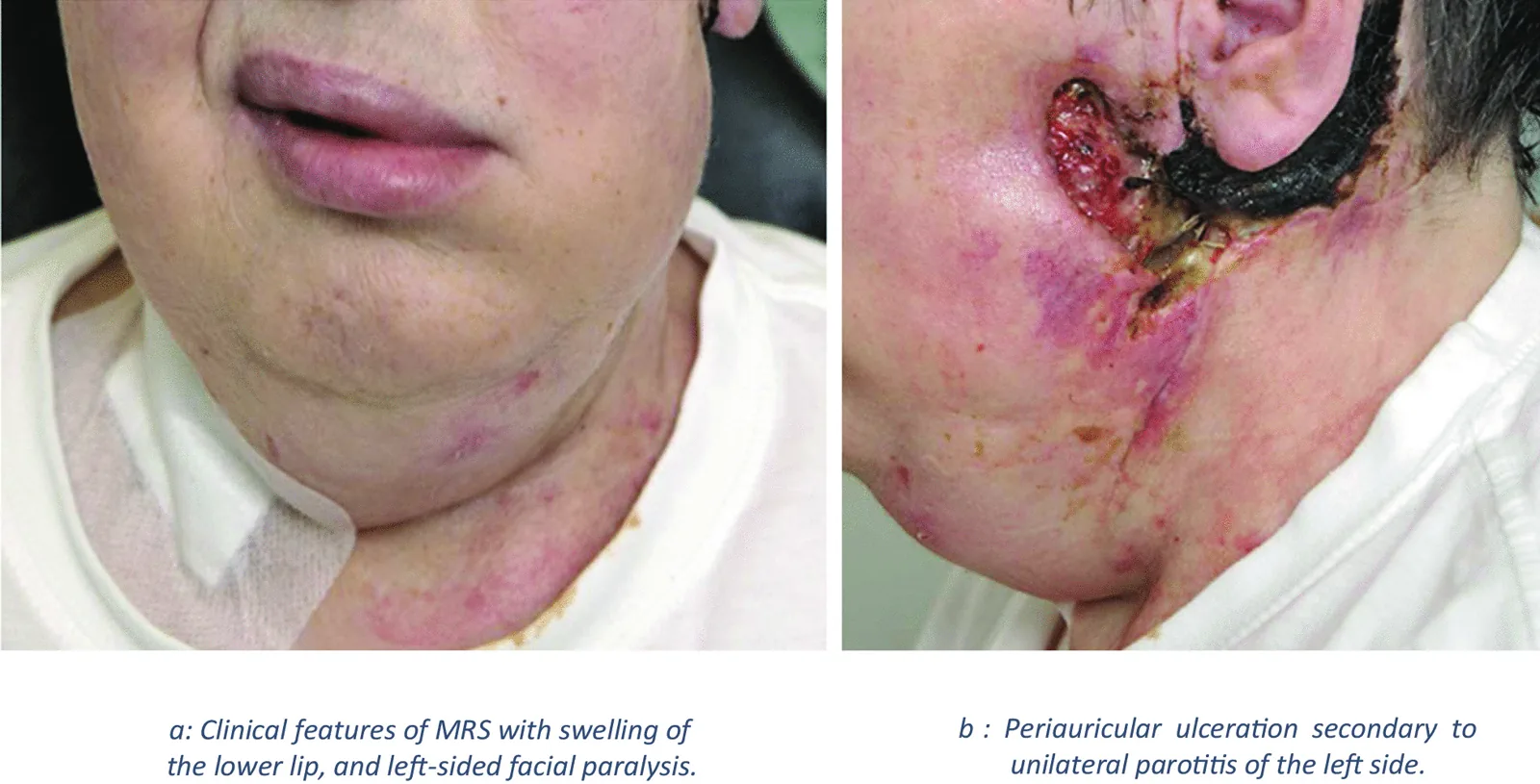
Das Melkersson-Rosenthal-Syndrom (MRS) ist eine nicht-verkäsende chronische granulomatöse neurokutane Erkrankung. Sie äußert sich in oralen und fazialen Ödemen, und eine Biopsie zeigt eine granulomatöse Entzündung. Es gehört zur Krankheitsgruppe der „orofazialen Granulomatose“.
Die historische Beschreibung dieser Erkrankung ist wie folgt. 1928 berichtete Ernst Melkersson erstmals detailliert über das Vorliegen einer rezidivierenden Gesichtslähmung und Ödeme. 1931 ergänzte Curt Rosenthal die Beschreibung der Lingua plicata (Faltenzunge), und 1949 verwendete Lüscher erstmals den Namen „Melkersson-Rosenthal-Syndrom“3).
Epidemiologisch wird geschätzt, dass es bei 0,08 % der Allgemeinbevölkerung auftritt2). Am häufigsten tritt es im Alter von 20 bis 30 Jahren auf, das Durchschnittsalter bei Beginn beträgt 39 Jahre (Spanne: 8–79 Jahre). Es besteht eine weibliche Prädominanz (etwa 3-fach).
Die klassische Trias besteht aus den folgenden drei Merkmalen.
Rezidivierendes orofaziales Ödem: insbesondere Lippenschwellung (granulomatöse Cheilitis)
Rezidivierende periphere Fazialisparese
Faltenzunge (Lingua plicata)
Alle drei Symptome treten nur bei 8–25 % der Fälle auf. Die meisten Patienten zeigen nur 1–2 Symptome, und die monosymptomatische Form wird manchmal als granulomatöse Cheilitis (Miescher-Cheilitis) beschrieben1).
Einige Forscher vermuten, dass diese Erkrankung ein Symptom der Sarkoidose oder des Morbus Crohn sein könnte, und ob es sich um eine eigenständige Erkrankung handelt, bleibt ungeklärt.
QKann die Diagnose nur gestellt werden, wenn alle drei Symptome vorhanden sind?
A
Die klassische Trias ist nur bei 8–25 % der Fälle vollständig. Die Diagnose ist auch möglich, wenn 1–2 Symptome durch Anamnese und körperliche Untersuchung bestätigt werden oder eine Biopsie eine granulomatöse Cheilitis zeigt. Eine definitive Diagnose ist selten, und eine langfristige Nachbeobachtung ist erforderlich.
J Casper, S Mohammad-Khani, J J Schmidt et al. Melkersson–Rosenthal syndrome in the context of sarcoidosis: a case report . Journal of Medical Case Reports. 2021 Oct 4; 15:488. Figure 2. PMCID: PMC8489098. License: CC BY.
Abbildung: Bild, das die Merkmale des Melkersson-Rosenthal-Syndroms zeigt
Die drei Elemente der Trias unterscheiden sich erheblich in Häufigkeit und Merkmalen.
Orofaziales Ödem
Häufigkeit: am höchsten, tritt bei 80–100 % der Fälle auf.
Typische Merkmale: einseitig, schmerzlos, nicht eindrückbar. Bevorzugt an der Oberlippe.
Verlauf: anfangs vorübergehend und intermittierend, kann aber später persistierend werden. Kann zu Gewebefibrose führen.
Differenzialdiagnose: ähnlich wie Angioödem, aber MRS ist persistierender und spricht nicht auf Antihistaminika an. Komplement normal.
Gesichtsnervenlähmung
Häufigkeit: 47–60 % der Fälle (bis zu 90 %). Einseitig oder beidseitig.
Verlauf: erste Episode erholt sich oft, aber mit Rezidiven verlängert sich die Dauer und kann permanent werden. Rezidivrate 3–11 %.
Beziehung zum Ödem: Kann einige Jahre vor oder nach dem orofazialen Ödem auftreten. Bei 13–50 % der Betroffenen treten Ödem und Fazialisparese gemeinsam auf.
Epidemiologie: Die Inzidenz der MRS-assoziierten Fazialisparese beträgt 0,36 Personen pro Jahr pro 100.000 Einwohner5).
Lingua plicata
Häufigkeit: 30–35 % der Fälle.
Beschaffenheit: Furchen von etwa 2 mm Tiefe und 15 mm Länge. Kann angeboren sein.
Diagnostische Bedeutung: Da sie auch bei 5 % der Normalbevölkerung vorkommt, ist sie allein kein diagnostisches Kriterium.
Als ophthalmologischer Befund wurde auch ein monosymptomatischer MRS berichtet, der nur ein Lidödem aufweist 3). Weitere begleitende ophthalmologische Befunde sind retrobulbäre Neuritis, Hornhauttrübung, Keratokonjunktivitis sicca und Blepharochalasis 3). Als systemische Komplikationen wurden Divertikulitis und Uveitis berichtet, was auf einen Zusammenhang mit Morbus Crohn und Sarkoidose hindeutet.
QWie unterscheidet man die Fazialisparese von der Bell-Lähmung?
A
Die Bell-Lähmung geht nicht mit einem orofazialen Ödem einher. Beim MRS hingegen wird bei 80–100 % der Fälle ein orofaziales Ödem beobachtet. Bei rezidivierender Fazialisparese mit orofazialem Ödem sollte aktiv an einen MRS gedacht werden.
Die Ätiologie ist unbekannt. Die folgenden Faktoren werden als beteiligt angesehen, es wurde jedoch kein starker Zusammenhang gefunden, der die Ursache des MRS erklärt.
Auslöser und Risikofaktoren werden wie folgt klassifiziert.
Hinsichtlich genetischer Faktoren wurde ein autosomal-dominanter Erbgang mit einem ursächlichen Gen auf dem kurzen Arm von Chromosom 9 (9p11) berichtet. Eine Mutation des ATP1 (FATP1)-Gens wurde in einer chinesischen Familie beschrieben, und auch eine heterozygote SLC27A1 (FATP1)-Mutation wurde berichtet, jedoch ohne endgültige Bestätigung3). In familiären Fällen von MRS wurde eine gefurchte Zunge bei Bruder, Sohn, Mutter und Schwester des Patienten festgestellt5).
Bei Infektionen können Virusinfektionen (EBV, VZV, CMV, HSV) Auslöser sein. Ein Fall eines Rückfalls nach 4 Jahren im Anschluss an eine COVID-19-Infektion wurde berichtet, was darauf hindeutet, dass die Aktivierung von Mastzellen sowohl an der Entzündungsreaktion von COVID-19 als auch an der Pathologie des MRS beteiligt sein könnte2).
Immunologisch wird eine Beteiligung der Th1-Typ-zellulären Immunantwort angenommen 1). Zu den Allergenen gehören glutenhaltige Lebensmittel, Schokolade, Zimt, Lebensmittelzusatzstoffe (Mononatriumglutamat), Zahnpasta, Metalle (Gold, Quecksilber, Kobalt, Zink) und Dentalamalgam 1).
Als verwandte Erkrankungen wird eine Überschneidung mit Morbus Crohn und Sarkoidose diskutiert. Bei der Diagnose der Sarkoidose wird MRS als eine der „anderen granulomatösen Hauterkrankungen“ aufgeführt, die ausgeschlossen werden sollten, und eine Unterscheidung zwischen beiden ist erforderlich.
QWenn in der Familie die gleichen Symptome auftreten, besteht dann ein genetischer Zusammenhang?
A
Bei MRS wird eine autosomal-dominante Vererbung vermutet, und es wurden Fälle von gefurchter Zunge oder Gesichtslähmung in Familien berichtet 5). Allerdings ist die klinische und genetische Vielfalt der Erkrankung groß, und die genetische Ursache ist noch nicht geklärt 3).
Die klinische Diagnose basiert auf der Krankengeschichte und der körperlichen Untersuchung. Das Vorliegen von mindestens zwei der drei Hauptmerkmale oder der Nachweis einer granulomatösen Cheilitis durch Biopsie gelten als ausreichend für die Diagnose 5). Eine definitive Diagnose ist selten, und eine langfristige Nachbeobachtung ist erforderlich.
Zur Differenzialdiagnose werden folgende Untersuchungen durchgeführt.
Serumkalzium und ACE-Aktivität : Normale Werte schließen eine Sarkoidose aus1)
Röntgen-Thorax und HRCT : Überprüfung auf hiläre Lymphadenopathie (Ausschluss einer Sarkoidose)
CT und MRT : Bestätigung einer Weichteilverdickung. MRT wird auch zur Beurteilung des Nervus facialis eingesetzt3)
Neuroelektrophysiologische Untersuchung : Bestätigung einer verminderten Amplitude des Aktionspotentials in der motorischen Neurographie des Nervus facialis5)
Wird eine Sarkoidose aufgrund von Augenveränderungen vermutet, sollten Blutuntersuchungen (Serum-ACE, löslicher IL-2-Rezeptor) und ein Röntgen-Thorax durchgeführt werden; gegebenenfalls ist eine Überweisung an einen Pneumologen oder Dermatologen erforderlich.
Weitere Differenzialdiagnosen sind Hypothyreose, Vena-cava-superior-Syndrom, Lymphangiom, Erysipel und Lymphom. Die Diagnose kann eine multidisziplinäre Zusammenarbeit zwischen Augenheilkunde, Dermatologie, Immunologie und Gastroenterologie erfordern.
Es gibt keine kurative Behandlung, und Rückfälle sind unvermeidlich. Spontanremissionen wurden berichtet, aber ohne Behandlung können die Symptomepisoden häufiger und länger andauern. Eine Standardtherapie ist nicht etabliert; der folgende abgestufte Ansatz wurde beschrieben.
Orale Steroide : Prednison (1 mg/kg/Tag, nach 10 Tagen ausschleichen). Hohe Remissionsrate, aber Rezidive5).
NSAR und Antihistaminika : werden als Initialtherapie eingesetzt.
Zweitlinientherapie und weitere
Antibiotika : Minocyclin 100 mg/Tag (wirksam in Kombination mit Steroiden)1)2). Auch Azithromycin-Pulstherapie (500 mg/Tag × 3 Tage/Woche) wurde versucht.
Immunsuppressiva und Biologika : Clofazimin, Methotrexat, Dapson, Sulfasalazin. Infliximab (basierend auf der Ähnlichkeit mit Morbus Crohn) 1).
Tacrolimus-Salbe 0,03 % : wirksam bei steroidrefraktärem Lidödem3).
Antivirale Medikamente (Aciclovir 400 mg alle 4 Stunden) können bei Fazialisparese zusammen mit Kortikosteroiden eingesetzt werden 5).
Dekompression des Gesichtsnervs : Indiziert bei persistierender oder rezidivierender Gesichtslähmung, die auf konservative Behandlung nicht anspricht. Erwägung ab House-Brackmann-Grad IV oder höher. Verbesserungen von Grad VI auf Grad II wurden berichtet (8 Monate postoperativ), und ein präventiver Effekt gegen zukünftige Rezidive wird vermutet 5). Auch eine verzögerte Dekompression nach 90 Tagen gilt als wirksam 5).
Cheiloplastik (Lippenplastik) : Wird bei therapieresistenter Lippenschwellung durchgeführt.
Helium-Neon-Laserbestrahlung: Es gibt Berichte, dass sie bei Patienten mit einer Erkrankungsdauer von weniger als 4 Jahren vorteilhaft ist.
Strahlentherapie: Eine konsistente Wirksamkeit wurde nicht nachgewiesen.
QIst eine Operation wirksam, wenn die medikamentöse Therapie nicht ausreicht?
A
Bei einer Gesichtslähmung, die auf eine konservative Behandlung nicht anspricht, ist die Dekompression des Gesichtsnervs eine wirksame Option. Es wurden dramatische Verbesserungen von einer schweren Lähmung (House-Brackmann-Grad VI) auf Grad II berichtet5), und es wird auch ein präventiver Effekt gegen zukünftige Rezidive vermutet. Es wurde keine kontrollierte Studie im Vergleich zur konservativen Behandlung durchgeführt, aber bei anhaltender Lähmung ab Grad IV sollte eine Operation in Betracht gezogen werden.
6. Pathophysiologie und detaillierter Krankheitsmechanismus
Die grundlegende Pathologie des MRS ist eine chronische granulomatöse Entzündung, die hauptsächlich aus nicht-verkäsenden Epitheloidzellgranulomen besteht. Histologisch finden sich mehrkernige Langhans-Riesenzellen, perivaskuläre mononukleäre Entzündungsinfiltrate, Lymphödem und Fibrose.
Immunologisch wurde die Beteiligung der Th1-Typ-zellvermittelten Immunantwort nachgewiesen 1). Die Aktivierung von Mastzellen fördert die Freisetzung von Zytokinen (IL-1, IL-6), was vermutlich lokale granulomatöse Entzündungen und Ödeme hervorruft 2).
Der Mechanismus der Fazialisparese ist die Kompression des Nervengewebes durch granulomatöse Infiltration und Ödem. Bei jedem Rezidiv wird der Nerv fortschreitend geschädigt, wodurch die Dauer der Lähmung verlängert wird 5).
Als genetische Grundlage werden Mutationen in der Region 9p11 und im FATP1 (SLC27A1)-Gen vermutet, jedoch ist die klinische und genetische Vielfalt der Erkrankung groß, sodass keine endgültige Schlussfolgerung gezogen werden konnte 3).
Es wird diskutiert, dass MRS, granulomatöse Cheilitis, Sarkoidose und Morbus Crohn ein kontinuierliches Spektrum bilden könnten 1). Diese Erkrankungen sind alle durch nicht-verkäsende Granulome aufgrund einer Th1-Immunantwort gekennzeichnet und weisen klinische und histopathologische Ähnlichkeiten auf.
7. Aktuelle Forschung und zukünftige Perspektiven (Berichte aus der Forschungsphase)
Talidere et al. (2021) berichteten über eine 51-jährige Frau, bei der nach einer COVID-19-Infektion nach 4 Jahren ein Rezidiv eines Melkersson-Rosenthal-Syndroms (MRS) auftrat2). Sie wies die klassische Trias aus Lippenschwellung, rechtseitiger Fazialisparese und Lingua plicata auf und besserte sich unter einer Therapie mit Hydroxychloroquin, Azithromycin und Steroiden. Es wurde diskutiert, dass Mastzellen eine gemeinsame Rolle in der Entzündungsreaktion von COVID-19 und MRS spielen könnten, und erstmals berichtet, dass eine COVID-19-Infektion ein Auslöser für ein MRS-Rezidiv sein kann.
Estacia et al. (2022) berichteten über eine Besserung bei einer 59-jährigen Frau mit isoliertem rechtem Lidödem (monosymptomatische MRS), die auf 5 intraläsionale Triamcinolon-Injektionen (über 1 Jahr) und orales Prednison nicht ansprach, nach Anwendung von Tacrolimus 0,03% Salbe3). Dies wird als neue lokale Therapie für steroidrefraktäres Lidödem diskutiert.
Ergebnisse der Gesichtsnervendekompression und familiäres MRS
Alencar et al. (2023) berichteten über eine deutliche Verbesserung (Grad II) nach einer Gesichtsnervendekompression beim dritten Anfall (linksseitige Lähmung Grad VI) bei einer 38-jährigen Frau mit familiärem MRS, acht Monate nach der Operation 5). Der Bruder, der Sohn, die Mutter und die Schwester der Patientin hatten eine gefurchte Zunge, und der Bruder hatte eine Vorgeschichte von Gesichtsnervenlähmung, was auf ein autosomal-dominantes Vererbungsmuster hindeutet. Dieser Fall, der auf eine konservative Behandlung (Prednison 60 mg/Tag, Aciclovir 400 mg) nicht angesprochen hatte, deutet auf die Wirksamkeit einer verzögerten Dekompression und einen möglichen präventiven Effekt gegen zukünftige Rezidive hin.
Als Kandidatengen für MRS wurde eine heterozygote Mutation von SLC27A1 (FATP1) berichtet, aber aufgrund der großen klinischen und genetischen Vielfalt ist es schwierig, die gesamte Erkrankung durch eine einzelne Genmutation zu erklären 3). Fortschritte in der Genforschung versprechen, die Pathogenese von MRS aufzuklären und therapeutische Ziele zu identifizieren.
Tummidi S, Nagendran P, Anthony ML, Ramani RJ, Shankaralingappa A, Gopinath H. Granulomatous cheilitis of Miescher: a rare entity. BMC women’s health. 2023;23(1):118. doi:10.1186/s12905-023-02280-9. PMID:36944970; PMCID:PMC10031989.
Taşlıdere B, Mehmetaj L, Özcan AB, Gülen B, Taşlıdere N. Melkersson-Rosenthal Syndrome Induced by COVID-19. The American journal of emergency medicine. 2021;41:262.e5-262.e7. doi:10.1016/j.ajem.2020.08.018. PMID:32829989; PMCID:PMC7428670.
Estacia CT, Gameiro Filho AR, da Silveira IBE, Gameiro RR, Barba ALSD. Melkersson-Rosenthal syndrome: a rare variant of the monosymptomatic form. GMS ophthalmology cases. 2022;12:Doc04. doi:10.3205/oc000191. PMID:35291587; PMCID:PMC8900159.
Aukerman E, List M, Avashia-Khemka N. Melkersson-Rosenthal syndrome of the vulva. JAAD case reports. 2022;27:35-37. doi:10.1016/j.jdcr.2022.07.006. PMID:35996444; PMCID:PMC9391513.
Alencar TN, Botelho MM, Carasek N, Bahmad F Jr. Surgical Treatment Outcome for Familial Melkersson-Rosenthal Syndrome. The American journal of case reports. 2023;24:e938670. doi:10.12659/AJCR.938670. PMID:36755481; PMCID:PMC9923774.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.