El síndrome de Miller Fisher (SMF) es una neuropatía autoinmune caracterizada por la tríada de oftalmoplejía externa aguda, ataxia y arreflexia. Se clasifica como un subtipo del síndrome de Guillain-Barré (SGB).
Charles Miller Fisher lo describió por primera vez en detalle en 1956. Se dice que James Collier ya había definido la tríada en 19321). El SMF forma parte del espectro del síndrome de anticuerpos anti-GQ1b, constituyendo un continuo de enfermedades junto con el SGB, la encefalitis del tronco encefálico de Bickerstaff y la oftalmoplejía aguda (tipo sin ataxia).
Epidemiología es la siguiente:
Prevalencia mundial: aproximadamente 1 por cada 1,000,000 de personas1)
Incidencia: 0,09 por 100.000 personas2)
Proporción del GBS: Alrededor del 5% en Occidente, 17–25% en Asia Oriental1)
Proporción de sexos: 2:1, más frecuente en hombres
Edad media de inicio: 40 años. Puede ocurrir a cualquier edad.
Tasa de recurrencia: 11–14%1)
El MFS suele tener un curso monofásico y autolimitado.
Q¿Qué tan raro es el síndrome de Miller Fisher?
A
La prevalencia mundial es de 1 por millón de personas y la incidencia es de 0.09 por 100,000, lo que lo convierte en una enfermedad rara1, 2). Sin embargo, en Asia oriental representa del 17 al 25% de los casos de GBS, una prevalencia mayor que el 5% aproximadamente en los países occidentales1).
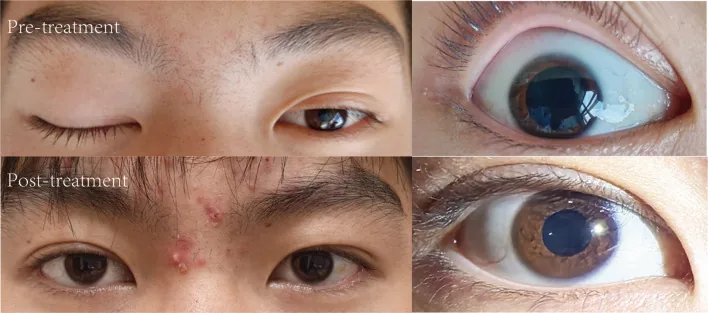
Juyuan Pan, Ningyu Zheng, Dan Yu et al. Unilateral ophthalmoplegia in anti-GQ1b antibody syndrome: case report and systematic literature review. Frontiers in Immunology. 2025 Oct 10; 16:1669821. Figure 1. PMCID: PMC12549634. License: CC BY.
Ilustración que muestra los tres síntomas principales del síndrome de Miller Fisher: oftalmoplejía, ataxia y arreflexia
La mayoría de los casos se presentan con diplopía o inestabilidad (ataxia) aproximadamente una semana después de una infección del tracto respiratorio superior. Los síntomas progresan durante 1 a 2 semanas después del inicio y luego tienden a remitir espontáneamente.
Los principales síntomas subjetivos se enumeran a continuación.
Trastorno de la marcha/inestabilidad: presente en el 32%
Anomalías sensoriales: presentes en el 14%
Otros: ptosis, alteración de la conciencia, debilidad de extremidades, disfagia, síntomas de parálisis bulbar, fotofobia, mareo, visión borrosa, cefalea, parálisis del nervio facial
A continuación se presentan los hallazgos clínicos representativos asociados con la tríada del MFS.
Oftalmoplejía
Oftalmoplejía externa: Generalmente bilateral y simétrica. La afectación unilateral se observa en el 27–31% de los casos4).
Oftalmoplejía interna (afectación pupilar): Aparece en aproximadamente la mitad de los casos. Se presenta con midriasis, pérdida del reflejo fotomotor y trastorno de la acomodación.
Ptosis: Puede observarse como un hallazgo fuera de la tríada.
Ataxia
Ataxia troncal y de extremidades: La gravedad varía. El 30% de los pacientes presenta ataxia lo suficientemente grave como para impedir la marcha independiente. Incapacidad para realizar la marcha en tándem.
Evolución tras el inicio: Tiende a remitir en aproximadamente 1 mes.
Pérdida de reflejos tendinosos
Pérdida de reflejos osteotendinosos profundos: Se considera relativamente característico de este síndrome. Los reflejos pueden conservarse en el 12–31% de los casos.
Evolución tras el inicio: La recuperación tiende a ser más lenta que la de la ataxia y la oftalmoplejía.
Orden de recuperación: La ataxia, la oftalmoplejía y la arreflexia se recuperan en ese orden1). La ataxia se resuelve en aproximadamente 1 mes, la oftalmoplejía externa en unos 3 meses, y la mayoría de los casos se resuelven sin secuelas en unos 6 meses.
Síndrome de superposición MFS/GBS: El 5.6–7.1% de los casos de MFS presentan debilidad en las extremidades2).
MFS incompleto: La oftalmoplejía aguda sin ataxia se reconoce como una forma del síndrome de anticuerpos anti-GQ1b5).
Q¿En qué orden se recuperan los síntomas del síndrome de Miller Fisher?
A
La recuperación progresa en el orden: ataxia (aproximadamente 1 mes) → oftalmoplejía (aproximadamente 3 meses) → arreflexia1). La mayoría de los pacientes se recuperan sin secuelas en unos 6 meses.
El mimetismo molecular es el principal mecanismo patogénico. Los lipooligosacáridos de los patógenos son estructuralmente similares al gangliósido humano GQ1b, induciendo anticuerpos anti-GQ1b de reacción cruzada.
Infección precedente y patógenos:
La infección del tracto respiratorio superior (76%) es la más frecuente. La infección gastrointestinal también se presenta en el 25%.
La mediana de tiempo desde la infección hasta la aparición de síntomas neurológicos es de 8 días.
Otros: Mycoplasma, citomegalovirus, virus de Epstein-Barr, virus varicela-zóster, VIH-1
Relacionado con vacunas: Se ha informado de aparición tras la vacuna contra la COVID-19 (BNT162b2, ChAdOx1, vacuna inactivada), la vacuna contra la gripe y la vacuna antineumocócica 5, 6, 7).
A continuación se resumen las principales infecciones y desencadenantes previos.
Tipo de desencadenante
Patógenos/preparados representativos
Infección bacteriana
C. jejuni, H. influenzae
Infección viral
Citomegalovirus, VEB, virus varicela-zóster
Vacunas
Vacuna contra la COVID-19, vacuna contra la influenza
Autoinmune/neoplásico
Enfermedad tiroidea, LES, tumores malignos (cáncer de pulmón, linfoma de Burkitt, etc.)3)
Factores de riesgo genéticos: Se ha sugerido una asociación entre HLA-DR2 y el SMR recurrente4).
Q¿Se puede desarrollar el síndrome de Miller Fisher después de un resfriado o una vacuna?
A
Se observa infección respiratoria superior previa en el 76% de los pacientes con SMR. También se han reportado múltiples casos después de la vacuna contra la COVID-19 (vacunas de ARNm e inactivadas) y la vacuna contra la influenza5, 6, 7). Sin embargo, algunos informes indican que el pronóstico del SMR posterior a la vacunación es favorable en todos los casos7).
El diagnóstico de MFS se basa en el diagnóstico clínico, confirmado por la tríada de oftalmoplejía, ataxia y arreflexia, y mediante la exclusión de otras enfermedades.
El anticuerpo IgG anti-GQ1b en suero es la prueba de diagnóstico auxiliar más importante.
Prueba
Sensibilidad
Especificidad
Anticuerpo IgG anti-GQ1b sérico
92%
97%1)
Anticuerpo anti-GQ1b en LCR
20%
100%1)
Los anticuerpos séricos anti-GQ1b son positivos en el 80–90% de los pacientes con MFS.
La prueba sérica es más sensible que el análisis del líquido cefalorraquídeo durante las primeras 3 semanas después del inicio.
Los títulos de anticuerpos se correlacionan con la gravedad de la oftalmoplejía.
Examen del líquido cefalorraquídeo: Muestra disociación albuminocitológica (albúmina elevada con recuento celular normal). Sin embargo, este hallazgo es común en enfermedades neuroinmunes.
Neuroimagen (RMN craneal): Generalmente normal. En raras ocasiones, pueden observarse anomalías inespecíficas en el cerebelo, pedúnculos cerebelosos medios o mesencéfalo, o realce del tronco encefálico.
Estudios de conducción nerviosa: A menudo normales.
El MFS es una enfermedad autolimitada; en casos con función respiratoria conservada, el tratamiento de apoyo (sintomático) puede ser suficiente. La mayoría de los casos remiten espontáneamente y tienen buen pronóstico.
Aunque no se ha establecido un tratamiento eficaz, se utilizan las siguientes inmunoterapias.
Dosis: 2 g/kg de peso corporal dividido en 5 días (400 mg/kg/día × 5 días) 3)
Eficacia: No se ha realizado un ECA para MFS. Los estudios retrospectivos muestran que no hay un impacto importante en los resultados, pero hay un informe de que acelera ligeramente el inicio de la recuperación de la oftalmoplejía (de 13.5 días a 12.0 días después del inicio).
Eficacia: Los informes de casos muestran éxito, pero los estudios retrospectivos no han demostrado un beneficio significativo. Se intenta junto con la terapia de inmunoadsorción para eliminar los anticuerpos anti-GQ1b.
Uso adicional de IVMP: Existen informes de casos que añaden terapia de pulsos de metilprednisolona (IVMP) en pacientes con recuperación insuficiente solo con IVIG8).
Es una enfermedad autolimitada con buen pronóstico, que a menudo vuelve a la normalidad en seis meses.
Ataxia: se resuelve en aproximadamente un mes
Oftalmoplejía externa: se resuelve en aproximadamente tres meses
Tiempo promedio de recuperación: aproximadamente 10 semanas
Síntomas residuales: presentes en hasta un tercio de los casos
Tasa de recurrencia: 3–14%; tasa de mortalidad: aproximadamente 4%
Q¿Puede el síndrome de Miller Fisher resolverse sin tratamiento?
A
La mayoría de los casos son enfermedades autolimitadas que remiten espontáneamente. La IVIG puede acelerar ligeramente la recuperación de la oftalmoplejía, pero los estudios retrospectivos no han demostrado un impacto significativo en el pronóstico. Sin embargo, si progresa a SGB o encefalitis del tronco encefálico de Bickerstaff, se recomienda IVIG o plasmaféresis 1).
Los lipooligosacáridos de patógenos como C. jejuni y H. influenzae son estructuralmente similares al gangliósido GQ1b, induciendo anticuerpos IgG anti-GQ1b de reacción cruzada después de la infección.
Gen cst-II de C. jejuni: polimorfismo Asn51 → anticuerpos anti-GQ1b → oftalmoplejía y ataxia / polimorfismo Thr51 → anticuerpos anti-GM1 y anti-GD1a → debilidad de extremidades (tipo SGB)
Asociación con la vacuna COVID-19: la proteína de espiga del SARS-CoV-2 se une a los gangliósidos a través del ácido siálico → mimetismo molecular 6)
GQ1b se expresa abundantemente en los siguientes sitios, y cada localización explica diferentes síntomas clínicos.
Regiones paranodales y terminales de los nervios oculomotores (III, IV, VI): Más abundante que en otros nervios craneales. Los anticuerpos anti-GQ1b son el mecanismo principal involucrado en la parálisis de los músculos extraoculares.
Células grandes de los ganglios de la raíz dorsal (neuronas del grupo Ia): El daño a las neuronas Ia causa ataxia sensorial y arreflexia debido a la alteración de la entrada sensorial.
Ganglio ciliar: Causa oftalmoplejía interna (trastornos pupilares y de acomodación).
Membrana presináptica de la unión neuromuscular (UNM): Anti-GQ1b se une a la UNM → liberación masiva de acetilcolina dependiente de complemento → bloqueo final de la transmisión neuromuscular. También ocurre destrucción mediada por complemento de los terminales axonales, sinapsis circundantes y células de Schwann.
GT1a se expresa abundantemente en los nervios glosofaríngeo y vago. En el SGB positivo para anti-GT1a, se han reportado parálisis de nervios craneales (parálisis ocular 57%, parálisis facial 57%, parálisis bulbar 70%), y el 39% requirió ventilación mecánica 8).
Liang et al. (2022) realizaron una revisión de alcance de 10 casos y resumieron las características clínicas del MFS después de la vacunación contra la COVID-19 7).
Edad media 63.5 años, 80% hombres, mediana de tiempo desde la vacunación hasta el inicio 13 días. Disociación albúmino-citológica en LCR 88.9%, anticuerpos antigangliósido positivos 62.5%. Todos los casos tuvieron buen pronóstico 7).
En el MFS después de la vacuna de ARNm (BNT162b2), la afinidad de la proteína espiga del SARS-CoV-2 por los gangliósidos a través del ácido siálico se destaca como mecanismo patogénico 6).
Se sabe que entre el 27% y el 31% de los pacientes con síndrome de anticuerpos anti-GQ1b presentan oftalmoplejía unilateral4).
En una revisión sistemática de 18 casos realizada por Pan et al. (2025), la mediana de edad fue de 31 años, con predominio masculino (13/18). El 44.4% de los niños presentó oftalmoplejía unilateral, frente a solo el 5.7% de los adultos. La mayoría se recuperó en un plazo de 3 meses4).
Existen informes de casos en los que se añadió IVMP en pacientes con recuperación prolongada tras IVIG sola8). Se ha sugerido un posible efecto de aceleración de la recuperación, pero el nivel de evidencia se limita a informes de casos.
La tasa de recurrencia se reporta entre el 11% y el 14%, y se ha sugerido una asociación con HLA-DR21). Las recurrencias pueden ser más leves (p. ej., solo oftalmoplejía) que el episodio inicial, y se han reportado casos de recuperación completa en un mes solo con tratamiento conservador1).
Los informes de MFS durante el embarazo son extremadamente raros. Se han reportado tanto el uso de IVIG como de plasmaféresis, sin informes de complicaciones perinatales9).
Ooi ST, Ahmad A, Yaakub A. Recurrent Miller Fisher Syndrome. Cureus. 2022;14(6):e26192. doi:10.7759/cureus.26192. PMID:35891880; PMCID:PMC9306407.
Bahk J, Yang W, Fishman J. Bilateral vocal cord paralysis in Miller Fisher syndrome/Guillain-Barre overlap syndrome and a review of previous case series. BMJ Case Rep. 2021;14:e240386. doi:10.1136/bcr-2020-240386.
Hakobyan N, Yadav R, Pokhrel A, Wasifuddin M, John MJ, Yadav S, et al. Miller-Fisher Syndrome Unveiled in the Presence of Cholangiocarcinoma. Cureus. 2023;15(11):e49016. doi:10.7759/cureus.49016. PMID:38111454; PMCID:PMC10727167.
Pan J, Zheng N, Yu D, Jiang H, Zhou Y. Unilateral ophthalmoplegia in anti-GQ1b antibody syndrome: case report and systematic literature review. Frontiers in immunology. 2025;16:1669821. doi:10.3389/fimmu.2025.1669821. PMID:41142776; PMCID:PMC12549634.
Abičić A, Adamec I, Habek M. Miller Fisher syndrome following Pfizer COVID-19 vaccine. Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology. 2022;43(3):1495-1497. doi:10.1007/s10072-021-05776-0. PMID:34817727; PMCID:PMC8611397.
Yamakawa M, Nakahara K, Nakanishi T, Nomura T, Ueda M. Miller Fisher Syndrome Following Vaccination against SARS-CoV-2. Internal medicine (Tokyo, Japan). 2022;61(7):1067-1069. doi:10.2169/internalmedicine.8851-21. PMID:35370249; PMCID:PMC9038467.
Liang H, Cao Y, Zhong W, Ma Z, Liu J, Chen H. Miller-Fisher syndrome and Guillain-Barre syndrome overlap syndrome following inactivated COVID-19 vaccine: Case report and scope review. Human vaccines & immunotherapeutics. 2022;18(6):2125753. doi:10.1080/21645515.2022.2125753. PMID:36315834; PMCID:PMC9746535.
Mitsuhashi S, Suzuki A, Hayashi K, Sato M, Nakaya Y, Takaku N, et al. Miller-Fisher Syndrome Following Influenza A Infection. Cureus. 2024;16(3):e56064. doi:10.7759/cureus.56064. PMID:38618457; PMCID:PMC11009552.
Ángel-Páez JA, Hurtado-Bugna S, Aragón-Mendoza RL, Altman-Restrepo M, Díaz-Yamal IJ, Centanaro-Meza GA. Miller Fisher syndrome treated with plasmapheresis during pregnancy: Case report and review of the literature. Revista colombiana de obstetricia y ginecologia. 2021;72(2):210-218. doi:10.18597/rcog.3611. PMID:34506707; PMCID:PMC8425356.
Copia el texto del artículo y pégalo en el asistente de IA que prefieras.
Artículo copiado al portapapeles
Abre un asistente de IA abajo y pega el texto copiado en el chat.