Idiopatica (54,6%)
Idiopatica: forma più comune, di causa sconosciuta 4)6). Benigna, con tendenza alla risoluzione spontanea.
Congenita: circa il 6% del totale. Nei neonati è coinvolta l’immaturità ipotalamica 2)6).

La sindrome di Arlecchino (Harlequin syndrome) è una sindrome autonomica causata da una disfunzione unilaterale del sistema nervoso simpatico. È caratterizzata da anidrosi e vasocostrizione (pallore) del viso e della parte superiore del corpo sul lato della lesione, e da sudorazione compensatoria e arrossamento controlaterale.
Fu descritta per la prima volta da Lance e Drummond nel 19881). Il nome deriva dalla maschera bicolore rossa e nera di Arlecchino, personaggio della commedia dell’arte italiana del XVI secolo6). Va notato che è una malattia completamente diversa dall’ittiosi arlecchino (una grave malattia cutanea ereditaria)6).
Il “segno di Arlecchino” (Harlequin sign) si riferisce al reperto fisico di arrossamento e sudorazione asimmetrici del viso, mentre la “sindrome di Arlecchino” è un concetto utilizzato in un contesto clinico più ampio, che include forme idiopatiche e congenite6).
In letteratura sono stati riportati circa 100 casi1)4); secondo una revisione di 108 casi di Guilloton e collaboratori, il 54,6% è idiopatico e il 45,4% secondario o iatrogeno4)6). La forma congenita rappresenta circa il 6% di tutti i casi6). È più frequente nelle donne e nella terza decade di vita1), con solo 37 casi pediatrici riportati1). Nei neonati si osserva con relativa frequenza a causa dell’immaturità funzionale dell’ipotalamo2).
Si tratta di una malattia completamente diversa. La sindrome di Harlequin è una malattia autonomica dovuta a disfunzione del sistema nervoso simpatico, mentre l’ittiosi arlecchino è una grave malattia cutanea ereditaria. I nomi delle malattie sono simili, ma la patologia, i sintomi e il trattamento sono completamente diversi6).
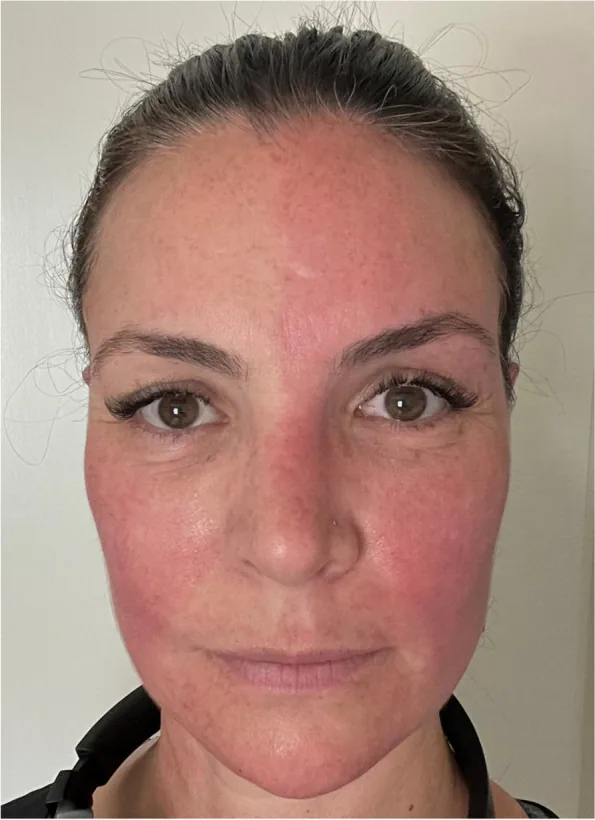
I sintomi compaiono in modo intermittente e possono manifestarsi a riposo senza una causa scatenante.
Il meccanismo patogenetico è l’interruzione dell’innervazione simpatica a livello pregangliare o postgangliare. Mentre la funzione simpatica sul lato colpito viene persa, il lato sano diventa compensatoriamente iperattivo, determinando reperti asimmetrici. Sono state riportate associazioni con emicrania e cefalee autonomiche trigeminali 1)3).
Idiopatica (54,6%)
Idiopatica: forma più comune, di causa sconosciuta 4)6). Benigna, con tendenza alla risoluzione spontanea.
Congenita: circa il 6% del totale. Nei neonati è coinvolta l’immaturità ipotalamica 2)6).
Secondaria (45,4%)
Lesioni compressive: tumore di Pancoast, tumore mediastinico, gozzo tiroideo (riportato un caso con gozzo multinodulare di 62×52×32 mm), dissezione carotidea 1)3)6)
Iatrogena: dopo simpaticectomia, dopo procedure anestesiologiche (es. blocco ESP), dopo posizionamento di catetere 1)5)
Malattie sistemiche: sindrome di Guillain-Barré, neuropatia diabetica, sclerosi multipla, infarto del tronco encefalico, siringomielia1)2)3)
La diffusione dell’anestetico nello spazio paravertebrale può bloccare temporaneamente il sistema simpatico, causando manifestazioni simili alla sindrome di Harlequin. È stato riportato un caso di insorgenza tardiva (dopo 5 ore) dopo un blocco paravertebrale a livello T2 con bupivacaina liposomiale5). Questa forma iatrogena di solito si risolve spontaneamente in 6-12 ore5).
La diagnosi è principalmente clinica, basata sui reperti clinici caratteristici6).
Gli esami di imaging sono importanti per escludere cause secondarie.
È necessaria la diagnosi differenziale con le seguenti malattie.
| Malattia | Punti di differenziazione |
|---|---|
| Sindrome di Ross | Triade: anidrosi estesa, assenza di riflessi tendinei profondi, pupilla di Adie |
| Sindrome di Horner | Solo miosi, ptosi palpebrale e anidrosi. Nessun rossore compensatorio controlaterale. |
| Sindrome di Holmes-Adie | Anomalie pupillari e assenza di riflessi tendinei profondi come caratteristiche principali |
La strategia terapeutica varia in base alla causa.
Nei casi secondari, il trattamento della malattia di base è prioritario6). La forma idiopatica è benigna e autolimitante; nella maggior parte dei casi è sufficiente consulenza e osservazione2)3).
Terapia farmacologica
Propranololo: beta-bloccante. Utilizzato per sopprimere sudorazione e rossore3).
Ossibutinina: farmaco anticolinergico. Ci si aspetta un effetto sull’iperidrosi3).
Procedure e interventi chirurgici
Iniezione di tossina botulinica: somministrazione locale nelle aree di iperidrosi compensatoria1).
Blocco del ganglio stellato: sollievo dei sintomi mediante blocco simpatico1)3).
Simpaticectomia toracica video-assistita (VATS): in una donna di 43 anni, i sintomi sono scomparsi completamente subito dopo l’intervento, senza recidive a 4 mesi4).
Nei casi idiopatici, la condizione è benigna e tende a risolversi spontaneamente, pertanto non sempre richiede trattamento2)3). Solo in caso di significativo disagio sociale o psicologico si possono considerare iniezioni di tossina botulinica, blocco del ganglio stellato o simpatectomia. Nei casi secondari, il trattamento della causa sottostante è prioritario.
La base della sindrome di Harlequin è un danno alla via simpatica a tre neuroni che innerva il volto e il tronco3).
La presenza o meno della sindrome di Horner varia a seconda del livello in cui si verifica la lesione lungo la via simpatica3)6).
| Sede della lesione | Sindrome di Horner associata | Caratteristiche cliniche |
|---|---|---|
| Lesione T1-T3 (simpatico oculare + fibre sudoripare e vasomotorie) | Sì | Sindrome di Harlequin + sindrome di Horner |
| Danno solo a T2-T3 | Nessuno | Sindrome di Harlequin isolata |
| Lesione postgangliare dopo il ganglio cervicale superiore | Presente (anidrosi mediale frontale) | Altre aree del viso possono essere risparmiate |
Quando il sistema simpatico sul lato affetto viene bloccato, l’attività simpatica sul lato sano aumenta relativamente, causando arrossamento e sudorazione compensatori1)6). I segni asimmetrici rispetto alla linea mediana sono dovuti a questo meccanismo. È stata anche suggerita la possibilità di una reinnervazione parasimpatica3).
La perdita della funzione del sistema nervoso simpatico sul lato affetto determina la rimozione dell’inibizione della vasodilatazione e della sudorazione su quel lato. Al contrario, sul lato sano non danneggiato, l’attività simpatica è relativamente aumentata, causando arrossamento e sudorazione compensatori1)6). La chiara differenza tra i due lati lungo la linea mediana riflette questa attività simpatica asimmetrica.
Tecik et al. (2025) hanno riportato che, in una donna di 43 anni con sindrome di Harlequin idiopatica persistente da 5 anni, è stata eseguita una simpatectomia VATS (toracoscopica), con completa risoluzione dei sintomi immediatamente dopo l’intervento e assenza di recidiva a 4 mesi di follow-up4). Nella revisione di Guilloton et al., il 54,6% dei casi era idiopatico4)6), suggerendo l’esistenza di un gruppo candidato al trattamento chirurgico.
Dalldorf et al. (2022) hanno riportato il primo caso di sindrome di Harlequin insorta con un ritardo di 5 ore dopo un blocco del piano erettore spinale (ESP block: bupivacaina liposomiale 133 mg + bupivacaina 0,25% 20 mL per lato) 5). Si tratta del primo rapporto di sindrome di Harlequin iatrogena correlata all’anestetico locale, che di solito si verifica entro 1 ora, ma in questo caso si è presentata tardivamente, e si ritiene che la modalità di insorgenza sia caratteristica delle formulazioni a rilascio prolungato (durata circa 72 ore). I sintomi si sono risolti spontaneamente entro la mattina successiva.
Strong et al. (2025) hanno riportato un caso di sindrome di Harlequin secondaria a gozzo multinodulare di 62×52×32 mm in una donna di 49 anni, sottoposta a tiroidectomia totale, ma i sintomi persistevano circa un anno dopo l’intervento 3). Questa osservazione è degna di nota in quanto mostra che le alterazioni croniche della via simpatica possono diventare irreversibili anche dopo la rimozione della lesione compressiva.