渦狀角膜(cornea verticillata)
頻率:出現在50%~80%的患者中
發病年齡:從6歲左右開始出現
部位:角膜上皮基底膜層
表現:灰白色淡色素沉著呈漩渦狀擴散至整個角膜
攜帶者:女性雜合子攜帶者也常見
法布瑞氏症是一種遺傳性代謝疾病,由於溶酶體水解酶α-半乳糖苷酶A(α-Gal A)缺乏,導致包括三己糖酰基鞘脂醇(Gb3)在內的鞘糖脂在全身細胞中蓄積。
由GLA基因(Xq22.1)突變引起X連鎖隱性遺傳。1898年由Anderson和Fabry分別報導2)。患病率估計約為1/10,000,但新生兒篩檢報告更高頻率(1:3,000至1:7,800)3)。
臨床表現大致分為經典型和遲發型(非經典型)。經典型中α-Gal A活性幾乎消失,從幼兒期開始出現多器官損害。遲發型中殘留酶活性,可能在成年期表現為心臟或腎臟的單器官受累1, 2)。
| 病型 | 發病時期 | 主要受影響器官 |
|---|---|---|
| 經典型 | 嬰幼兒至學齡期 | 多器官(全身) |
| 晚發型 | 成年期 | 心臟和腎臟 |
是的,這是一種X染色體隱性遺傳疾病。男性患者的全部女兒皆為帶因者,兒子則不會遺傳。女性帶因者因X染色體不活化模式不同,可能從無症狀到重症表現各異。如果您的家人患有此病,建議進行遺傳諮詢。
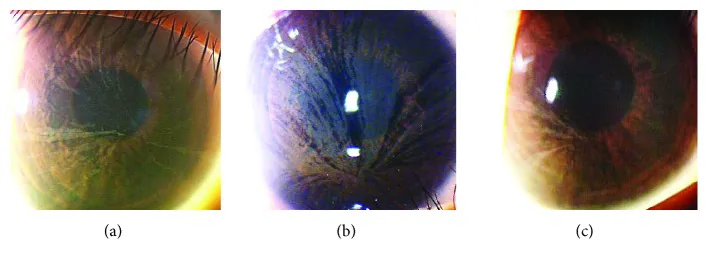
角膜渦狀混濁導致的視力下降很少見。多數情況下,眼部症狀的主觀主訴很少。
全身症狀包括四肢末端的灼痛(肢端感覺異常)是最早的症狀。出汗減少(低汗症)和胃腸道症狀也在兒童期出現。
渦狀角膜(cornea verticillata)
頻率:出現在50%~80%的患者中
發病年齡:從6歲左右開始出現
部位:角膜上皮基底膜層
表現:灰白色淡色素沉著呈漩渦狀擴散至整個角膜
攜帶者:女性雜合子攜帶者也常見
法布瑞氏白內障
血管系統變化
結膜血管:可見迂曲和擴張
視網膜血管:特徵性雙眼主幹靜脈迂曲
微動脈瘤:可見於結膜和視網膜
遲發型或心臟局限型可能缺乏典型的角膜和皮膚表現。據報導,一例攜帶W162C突變的心臟型法布里病患者,眼科評估未發現角膜渦狀混濁或血管角化瘤1)。
法布瑞氏症是由GLA基因突變引起的。已報告超過1000種突變,突變類型不同,臨床表現差異很大2)。
由於是X連鎖隱性遺傳病,男性(半合子)症狀往往較重。女性攜帶者(雜合子)因X染色體不活化偏斜,也可表現出從無症狀到接近經典型的嚴重症狀等多種臨床表現1)。約1%的女性攜帶者會出現症狀,角膜混濁對男女篩查均有幫助。
主要風險因素是法布瑞氏症的家族史。對於原因不明的肢端疼痛、年輕型中風、原因不明的心肌肥厚或原因不明的腎臟損害患者,需將本病納入鑑別診斷2, 4)。
裂隙燈顯微鏡檢查最為重要。渦狀角膜是特徵性發現,為懷疑本病的契機。也需確認有無白內障(後囊下車軸狀混濁)。非典型病例可能缺乏眼科表現1),因此不能因無表現而排除本病。
| 檢查項目 | 診斷意義 |
|---|---|
| α-Gal A活性 | 男性確定 |
| Lyso-Gb3 | 生物標記 |
| GLA基因分析 | 突變鑑定 |
男性可透過測量α-Gal A酶活性進行確診。女性攜帶者因X染色體去活化的影響,酶活性可能顯示正常範圍,因此需要進行基因分析2)。血漿Lyso-Gb3是一種診斷靈敏度高的生物標記,對女性篩檢也有用4)。
渦狀角膜最重要的鑑別診斷是**胺碘酮(抗心律不整藥)**引起的藥物性角膜沉積症。服用胺碘酮產生的混濁與法布瑞氏症的渦狀角膜極為相似。確認用藥史是鑑別的關鍵。
此外,還需要與氯喹、吲哚美辛等藥物引起的角膜沉積,以及其他導致角膜渦狀混濁的疾病進行鑑別。
法布瑞氏症的根本治療是酵素替代療法(ERT)。每兩週靜脈滴注阿加糖酶α(Replagal®)或阿加糖酶β(Fabrazyme®)。ERT有望減輕肢端疼痛、延緩腎功能下降、抑制心臟肥大的進展。早期開始治療對於抑制器官損傷的進展至關重要。
米加司他(Galafold®)是一種口服藥物,適用於具有可治療突變的患者。它能穩定變性的α-Gal A蛋白質的立體結構,促進其運送到溶酶體。
渦狀角膜病變無需特殊治療。如果白內障進展並影響視功能,則可考慮白內障手術。
酵素替代療法(ERT)可抑制腎臟、心臟和神經症狀的進展,但通常無法改善角膜的漩渦狀沉積。然而,由於角膜表現對視力影響不大,因此無需積極治療。如果白內障影響視力,則進行手術。
法布瑞氏症中,GLA基因突變導致α-半乳糖苷酶A活性降低或喪失。結果,該酶本應分解的Gb3及其去醯基形式Lyso-Gb3在溶酶體中積累。積累的Gb3引起細胞功能障礙、發炎級聯反應激活和氧化壓力,導致進行性多器官損傷。
在角膜中,Gb3積聚在角膜緣的基底幹細胞中。這些細胞在離心遷移過程中形成漩渦狀沉積模式(角膜渦狀混濁)。沉積發生在角膜上皮基底膜水平。
在晶狀體中,Gb3積聚在上皮細胞和皮質纖維中,導致後囊下出現車軸狀混濁。此發現稱為法布瑞氏白內障。
血管內皮細胞和平滑肌細胞中的Gb3積聚導致結膜和視網膜血管迂曲擴張。在視網膜中,表現為雙眼主幹靜脈迂曲。全身性方面,腎絲球足細胞和心肌細胞中的積聚導致腎損傷和心臟肥大。
除了傳統的ERT,法布瑞氏症的多種新型治療方法正在研究中。
基因治療通過腺相關病毒(AAV)載體導入GLA基因,產生內源性α-Gal A,有望成為一種根治性治療方法。
**基質合成抑制療法(SRT)**是一種抑制Gb3本身合成的途徑,與ERT不同,它正在開發為一種無需靜脈給藥的口服藥物。
擴大新生兒篩檢也是一個重要課題。早期診斷和早期治療介入可能改善長期預後3)。
關於表現型多樣性的知識也在累積。據報導,即使具有相同的GLA突變,也可能出現從經典型到器官局限型的不同臨床表現1),並且女性攜帶者中的X染色體不活化模式不能預測表現型。在以神經系統症狀為主訴的非經典型法布瑞氏症病例報告中,有透過詳細檢查腦白質病變而診斷出法布瑞氏症的例子2)。