Morbus Fabry ist eine erbliche Stoffwechselerkrankung, die durch einen Mangel an α-Galactosidase A (α-Gal A), einem lysosomalen Hydrolyseenzym, verursacht wird und zur Ansammlung von Sphingoglykolipiden, einschließlich Globotriaosylceramid (Gb3), in den Zellen des gesamten Körpers führt.
Sie wird X-chromosomal rezessiv vererbt, verursacht durch Mutationen im GLA-Gen (Xq22.1). Sie wurde 1898 unabhängig voneinander von Anderson und Fabry beschrieben 2). Die Prävalenz wird auf etwa 1 von 10.000 Personen geschätzt, aber Neugeborenen-Screenings berichten über eine höhere Häufigkeit (1:3.000 bis 1:7.800) 3).
Das klinische Bild wird in klassische Form und Spätform (nicht-klassisch) unterteilt. Bei der klassischen Form ist die α-Gal A-Aktivität nahezu aufgehoben, und bereits im Kindesalter treten Multiorganstörungen auf. Bei der Spätform besteht eine Restenzymaktivität, und die Erkrankung kann sich im Erwachsenenalter als isolierte Herz- oder Nierenerkrankung manifestieren 1, 2).
Krankheitstyp
Beginn
Hauptbetroffene Organe
Klassisch
Kleinkind- bis Schulalter
Multi-Organ (systemisch)
Spätmanifestation
Erwachsenenalter
Herz und Nieren
QIst der Morbus Fabry erblich?
A
Ja, es handelt sich um eine X-chromosomal rezessiv vererbte Erkrankung. Alle Töchter eines männlichen Patienten sind Konduktorinnen, während Söhne nicht betroffen sind. Weibliche Konduktorinnen können je nach X-Inaktivierungsmuster ein breites Spektrum von asymptomatischen bis hin zu schweren Symptomen aufweisen. Wenn in Ihrer Familie jemand an dieser Erkrankung leidet, wird eine genetische Beratung empfohlen.
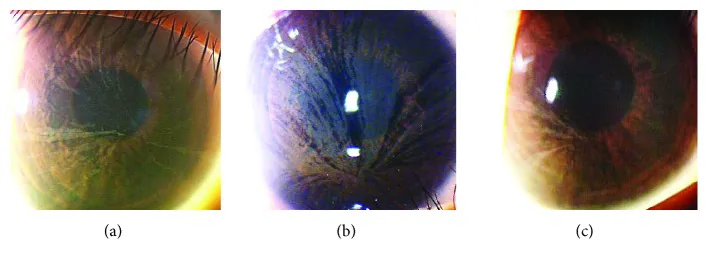
Yasuhito Ikegawa, Atsushi Shiraishi, Yasuhito Hayashi, Akiyoshi Ogimoto, et al. In Vivo Confocal Microscopic Observations of Vortex Keratopathy in Patients with Amiodarone-Induced Keratopathy and Fabry Disease 2018 Mar 21 J Ophthalmol. 2018 Mar 21; 2018:5315137 Figure 4. PMCID: PMC5884153. License: CC BY.
Spaltlampenmikroskopische Befunde von drei Patienten mit Morbus Fabry. (a) Befunde der 32-jährigen Mutter. (b) Befunde der 8-jährigen Tochter. (c) Befunde der 4-jährigen Tochter.
Eine Sehverschlechterung durch die spiralförmige Hornhauttrübung ist selten. In vielen Fällen gibt es wenige subjektive Beschwerden durch Augensymptome.
Zu den systemischen Symptomen gehören brennende Schmerzen in den Extremitäten (Akroparesthesie) als frühestes Symptom. Vermindertes Schwitzen (Hypohidrose) und gastrointestinale Symptome treten ebenfalls bereits im Kindesalter auf.
Lokalisation: auf Höhe der Basalmembran des Hornhautepithels
Befund: blassgraue Pigmentablagerungen, die sich wirbelförmig über die gesamte Hornhaut ausbreiten
Träger: auch bei weiblichen heterozygoten Trägerinnen häufig beobachtet
Fabry-Katarakt
Lokalisation : hintere Kapsel und Linsenrinde
Befund : speichenartige Trübungen (spoke-like) sind charakteristisch
Auswirkung auf das Sehvermögen : in der Regel gering, aber in fortgeschrittenen Fällen kann eine Kataraktoperation indiziert sein
Gefäßveränderungen
Bindehautgefäße : Schlängelung und Erweiterung
Netzhautgefäße: Charakteristisch ist eine beidseitige Schlängelung der Hauptvenen
Mikroaneurysmen: Können in der Bindehaut und Netzhaut auftreten
Bei spätmanifesten oder kardialen Formen können die typischen Hornhaut- und Hautbefunde fehlen. Bei einem Fall von kardialem Morbus Fabry mit W162C-Mutation wurden weder eine Cornea verticillata noch Angiokeratome in der ophthalmologischen Untersuchung festgestellt1).
QBeeinträchtigen die Hornhautwirbel die Sehkraft?
A
Die Cornea verticillata ist eine Ablagerung in den oberflächlichen Schichten des Hornhautepithels und beeinträchtigt in der Regel nicht die Sehkraft. Sie wird oft erstmals bei der Spaltlampenuntersuchung entdeckt, und der Patient selbst bemerkt sie kaum. Bei Fortschreiten eines Katarakts kann es jedoch zu einer Sehverschlechterung kommen.
Die Ursache des Morbus Fabry ist eine Mutation im GLA-Gen. Es wurden über 1000 verschiedene Mutationen beschrieben, und das klinische Bild variiert stark je nach Mutationstyp2).
Da es sich um einen X-chromosomal rezessiven Erbgang handelt, sind Männer (Hemizygote) häufiger schwer betroffen. Auch weibliche Konduktorinnen (Heterozygote) zeigen aufgrund einer X-Chromosom-Inaktivierung ein breites klinisches Spektrum von asymptomatisch bis hin zu schweren, klassischen Verläufen1). Etwa 1% der weiblichen Konduktorinnen entwickeln Symptome, und der Nachweis einer Hornhauttrübung ist für das Screening bei beiden Geschlechtern nützlich.
Der Hauptrisikofaktor ist eine positive Familienanamnese für Morbus Fabry. Bei Patienten mit unklaren akralen Schmerzen, juvenilem Schlaganfall, unklarer Kardiomegalie oder unklarer Niereninsuffizienz sollte diese Erkrankung in die Differenzialdiagnose einbezogen werden2, 4).
Die Spaltlampenmikroskopie ist am wichtigsten. Die Cornea verticillata ist ein charakteristischer Befund und gibt Anlass, an diese Erkrankung zu denken. Überprüfen Sie auch das Vorhandensein einer Katarakt (hintere subkapsuläre speichenförmige Trübung). Bei nicht-klassischen Fällen können augenärztliche Befunde fehlen 1); das Fehlen von Befunden sollte diese Erkrankung nicht ausschließen.
Bei Männern kann die definitive Diagnose durch Messung der α-Gal A-Enzymaktivität gestellt werden. Bei weiblichen Trägerinnen kann die Enzymaktivität aufgrund der X-Inaktivierung im Normalbereich liegen, sodass eine genetische Analyse erforderlich ist 2). Plasma-Lyso-Gb3 ist ein Biomarker mit hoher diagnostischer Sensitivität und auch für das Screening bei Frauen nützlich 4).
Die wichtigste Differentialdiagnose der Cornea verticillata ist die medikamentöse Hornhautablagerung durch Amiodaron (Antiarrhythmikum). Die durch die Einnahme von Amiodaron verursachten Trübungen ähneln der Cornea verticillata bei Morbus Fabry sehr. Die Überprüfung der Medikamentenanamnese ist der Schlüssel zur Differentialdiagnose.
Darüber hinaus ist eine Abgrenzung zu anderen medikamentösen Hornhautablagerungen wie durch Chloroquin oder Indometacin sowie zu anderen Erkrankungen, die eine Cornea verticillata verursachen, erforderlich.
Die kausale Behandlung des Morbus Fabry ist die Enzymersatztherapie (enzyme replacement therapy: ERT). Agalsidase alfa (Replagal®) oder Agalsidase beta (Fabrazyme®) werden alle zwei Wochen intravenös infundiert. Die ERT kann die Schmerzen in den Extremitäten lindern, das Fortschreiten der Nierenfunktionsverschlechterung verlangsamen und die Progression der Herzhypertrophie hemmen. Ein frühzeitiger Beginn ist wichtig, um das Fortschreiten von Organschäden zu verlangsamen.
Migalastat (Galafold®) ist ein orales Medikament, das für Patienten mit amenablen Mutationen indiziert ist. Es stabilisiert die räumliche Struktur des denaturierten α-Gal A-Proteins und fördert dessen Transport zum Lysosom.
Für die wirbelförmige Hornhauttrübung ist keine spezifische Behandlung erforderlich. Bei fortschreitendem Katarakt, der die Sehfunktion beeinträchtigt, ist eine Kataraktoperation indiziert.
QVerbessert die Behandlung die Hornhautsymptome?
A
Die Enzymersatztherapie (ERT) kann das Fortschreiten von Nieren-, Herz- und Nervensymptomen verlangsamen, verbessert jedoch in der Regel nicht die wirbelförmigen Hornhautablagerungen. Da diese Hornhautbefunde die Sehkraft nicht wesentlich beeinträchtigen, ist keine aggressive Behandlung erforderlich. Wenn der Graue Star die Sehkraft beeinträchtigt, wird eine Operation durchgeführt.
6. Pathophysiologie und detaillierter Krankheitsmechanismus
Bei Morbus Fabry führt eine Mutation im GLA-Gen zu einer verminderten oder fehlenden Aktivität der α-Gal A. Infolgedessen akkumulieren Gb3 und seine deacylierte Form Lyso-Gb3, die normalerweise von diesem Enzym abgebaut werden sollten, in den Lysosomen. Die Ansammlung von Gb3 verursacht Zellfunktionsstörungen, Aktivierung von Entzündungskaskaden und oxidativen Stress, was zu fortschreitenden Multiorganschäden führt.
In der Hornhaut akkumuliert Gb3 in den basalen Stammzellen des Limbus. Bei der zentrifugalen Migration dieser Zellen entsteht ein wirbelförmiges Ablagerungsmuster (Cornea verticillata). Die Ablagerungen treten auf Höhe der Basalmembran des Hornhautepithels auf.
In der Linse akkumuliert Gb3 in Epithelzellen und kortikalen Fasern, was zu einer speichenartigen Trübung unter der hinteren Kapsel führt. Dies wird als Fabry-Katarakt bezeichnet.
Die Akkumulation von Gb3 in vaskulären Endothelzellen und glatten Muskelzellen verursacht eine Schlängelung und Erweiterung der Bindehaut- und Netzhautgefäße. In der Netzhaut wird sie als bilaterale Schlängelung der Hauptvenen beobachtet. Systemisch führt die Akkumulation in renalen glomerulären Podozyten und Kardiomyozyten zu Nierenschäden und Herzhypertrophie.
Neben der konventionellen ERT werden mehrere neue Therapieansätze für die Fabry-Krankheit untersucht.
Die Gentherapie, bei der das GLA-Gen mittels eines Adeno-assoziierten Virus (AAV)-Vektors eingeführt wird, um endogen α-Gal A zu produzieren, wird als vielversprechende kurative Behandlung angesehen.
Substratreduktionstherapie (SRT) ist ein Ansatz, der die Synthese von Gb3 selbst hemmt. Im Gegensatz zur ERT wird sie als orales Medikament entwickelt, das keine intravenöse Verabreichung erfordert.
Die Ausweitung des Neugeborenen-Screenings ist ebenfalls ein wichtiges Thema. Eine frühzeitige Behandlung durch frühzeitige Diagnose könnte die Langzeitprognose verbessern3).
Auch das Wissen über die phänotypische Vielfalt nimmt zu. Es wurde berichtet, dass selbst bei derselben GLA-Mutation unterschiedliche klinische Bilder von der klassischen bis zur organspezifischen Form auftreten können1) und dass das X-Chromosom-Inaktivierungsmuster bei weiblichen Trägerinnen den Phänotyp nicht vorhersagt. In Fallberichten über nicht-klassischen Morbus Fabry mit neurologischen Symptomen als Hauptbeschwerde gab es Fälle, bei denen die Diagnose Morbus Fabry nach einer gründlichen Untersuchung von Hirnweißläsionen gestellt wurde2).
Furia A, Ditaranto R, Biagini E, et al. Fabry disease in W162C mutation: a case report of two patients and a review of literature. BMC Neurol. 2024;24:113. PMCID: PMC10996216. doi:10.1186/s12883-024-03540-3.
Ferreira Tátá C, Massas M, Pinto F, Caçador N, Silva AL. Fabry Disease: A Atypical Presentation. Cureus. 2021;13(10):e18708. doi:10.7759/cureus.18708. PMID:34790463; PMCID:PMC8582620.
Starcea IM, Bodescu Amancei Ionescu L, Lazaruc TI, Lupu VV, Bogos RA, Ioniuc I, et al. Palm-Plant Pain, Sign of a Severe Systemic Disease? Case Report and Review of Literature. Genes. 2023;14(2). doi:10.3390/genes14020516. PMID:36833443; PMCID:PMC9957027.
Yanfang W, Juanjuan H, Shengli Z, Lei Y, Fei G. Case report and literature review: Fabry disease misdiagnosing as polymyalgia rheumatica. Medicine. 2023;102(44):e34630. doi:10.1097/MD.0000000000034630. PMID:37933054; PMCID:PMC10627660.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.