Fabry hastalığı, lizozomal hidrolaz enzimi α-galaktosidaz A (α-Gal A) eksikliği nedeniyle globotriaosilseramid (Gb3) başta olmak üzere glikosfingolipidlerin tüm vücut hücrelerinde biriktiği kalıtsal bir metabolik hastalıktır.
GLA genindeki (Xq22.1) mutasyon nedeniyle X’e bağlı resesif kalıtım gösterir. İlk kez 1898’de Anderson ve Fabry tarafından bağımsız olarak bildirilmiştir 2). Görülme sıklığı yaklaşık 10.000’de 1 olarak tahmin edilmekle birlikte, yenidoğan taramalarında daha yüksek sıklık (1:3.000 ila 1:7.800) bildirilmiştir 3).
Klinik tablo klasik ve geç başlangıçlı (klasik olmayan) olarak ikiye ayrılır. Klasik tipte α-Gal A aktivitesi neredeyse tamamen kaybolmuştur ve çocukluktan itibaren çoklu organ hasarı ortaya çıkar. Geç başlangıçlı tipte ise kalan enzim aktivitesi vardır ve erişkin dönemde kalp veya böbrek gibi tek organ hasarı olarak ortaya çıkabilir 1, 2).
Hastalık tipi
Başlangıç zamanı
Başlıca etkilenen organlar
Klasik tip
Bebeklik-okul çağı
Çoklu organ (tüm vücut)
Geç başlangıçlı
Yetişkinlik
Kalp ve böbrek
QFabry hastalığı kalıtsal mıdır?
A
Evet, X’e bağlı resesif kalıtım gösteren bir hastalıktır. Erkek hastaların tüm kızları taşıyıcı olur, oğullarına geçmez. Kadın taşıyıcılar, X kromozomu inaktivasyon paternine bağlı olarak asemptomatikten şiddetliye kadar geniş bir semptom yelpazesi gösterebilir. Ailenizde bu hastalığa sahip biri varsa, genetik danışmanlık almanız önerilir.
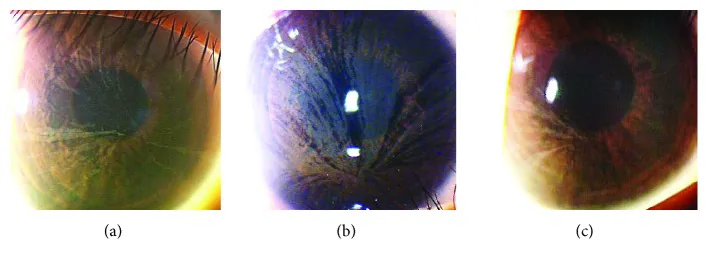
Yasuhito Ikegawa, Atsushi Shiraishi, Yasuhito Hayashi, Akiyoshi Ogimoto, et al. In Vivo Confocal Microscopic Observations of Vortex Keratopathy in Patients with Amiodarone-Induced Keratopathy and Fabry Disease 2018 Mar 21 J Ophthalmol. 2018 Mar 21; 2018:5315137 Figure 4. PMCID: PMC5884153. License: CC BY.
Fabry hastalığı olan üç hastanın yarık lamba biyomikroskopi bulguları. (a) 32 yaşındaki annenin bulguları. (b) 8 yaşındaki kızının bulguları. (c) 4 yaşındaki kızının bulguları.
Korneadaki sarmal bulanıklığa bağlı görme azalması nadirdir. Çoğu durumda, oküler semptomlara bağlı sübjektif şikayetler azdır.
Sistemik semptomlar arasında ekstremitelerde yanıcı ağrı (akroparestezi) en erken belirtidir. Terleme azalması (hipohidroz) ve gastrointestinal semptomlar da çocukluktan itibaren ortaya çıkar.
Başlangıç yaşı: Yaklaşık 6 yaşından itibaren fark edilir
Yerleşim: Kornea epitel bazal membran seviyesinde
Bulgular: Grimsi soluk pigment birikintilerinin tüm korneada sarmal şekilde yayılması
Taşıyıcılar: Kadın heterozigot taşıyıcılarda da yüksek sıklıkta gözlenir
Fabry Kataraktı
Yer: Arka subkapsüler ve lens korteksi
Bulgular: Tekerlek teli benzeri (spoke-like) opasiteler karakteristiktir
Görme üzerine etkisi: Genellikle hafiftir ancak ilerlemiş vakalarda katarakt cerrahisi endikasyonu oluşur
Vasküler Değişiklikler
Konjonktival damarlar: Kıvrımlılık ve genişleme görülür
Retina damarları: İki taraflı ana venlerde kıvrımlılık karakteristiktir
Mikroanevrizmalar: Konjonktiva ve retinada görülebilir
Geç başlangıçlı veya kardiyak sınırlı tipte, kornea ve derinin tipik bulguları eksik olabilir. W162C mutasyonuna sahip kardiyak Fabry hastalığı olan bir vakada, oftalmolojik değerlendirmede ne kornea verticillata ne de anjiyokeratom saptanmadığı bildirilmiştir1).
Kornea verticillata, kornea epitelinin yüzeysel tabakasında oluşan bir birikimdir ve genellikle görme keskinliğini etkilemez. Genellikle yarık lamba muayenesinde ilk kez fark edilir ve hastanın kendisi tarafından nadiren fark edilir. Ancak katarakt ilerlerse görme azalması meydana gelebilir.
Fabry hastalığının nedeni GLA genindeki mutasyondur. 1000’den fazla mutasyon türü rapor edilmiştir ve mutasyon türüne bağlı olarak klinik tablo büyük ölçüde değişir 2).
X’e bağlı resesif kalıtım nedeniyle erkekler (hemizigot) daha ağır seyretmeye eğilimlidir. Kadın taşıyıcılar (heterozigot) da X kromozomu inaktivasyonundaki dengesizlik nedeniyle asemptomatikten klasik tipe yakın ağır vakalara kadar çeşitli klinik tablolar gösterir 1). Kadın taşıyıcıların yaklaşık %1’inde semptomlar görülür ve kornea bulanıklığının tespiti hem erkekler hem de kadınlar için taramada faydalıdır.
Ana risk faktörü Fabry hastalığı aile öyküsüdür. Nedeni bilinmeyen ekstremite uçlarında ağrı, genç yaşta inme, nedeni bilinmeyen kalp büyümesi veya nedeni bilinmeyen böbrek yetmezliği olan hastalarda bu hastalık ayırıcı tanıda düşünülmelidir 2, 4).
Yarık lamba biyomikroskopisi en önemlisidir. Kornea verticillata karakteristik bir bulgudur ve bu hastalıktan şüphelenmeye yol açar. Katarakt (arka subkapsüler aksiyel opasite) varlığı da kontrol edilir. Klasik olmayan vakalarda göz bulguları olmayabilir1), bu nedenle bulgu olmaması hastalığı dışlamamalıdır.
Erkeklerde α-Gal A enzim aktivitesinin ölçümü ile kesin tanı konulabilir. Kadın taşıyıcılarda X inaktivasyonu nedeniyle enzim aktivitesi normal aralıkta görülebileceğinden genetik analiz gerekir2). Plazma Lyso-Gb3, yüksek tanısal duyarlılığa sahiptir ve kadınların taranmasında da yararlı bir biyobelirteçtir4).
Kornea verticillata’nın en önemli ayırıcı tanısı, amiodaron (antiaritmik ilaç) kaynaklı ilaca bağlı kornea birikintileridir. Amiodaron kullanımına bağlı oluşan bulanıklıklar, Fabry hastalığındaki kornea verticillata’ya çok benzer bir görünüm sergiler. İlaç öyküsünün sorgulanması ayırıcı tanıda anahtardır.
Ayrıca klorokin, indometazin gibi ilaçlara bağlı kornea birikintileri ve cornea verticillata’ya neden olan diğer hastalıklarla ayırıcı tanı yapılması gerekir.
Fabry hastalığının temel tedavisi enzim replasman tedavisidir (enzyme replacement therapy: ERT). Agalsidaz alfa (Replagal®) veya agalsidaz beta (Fabrazyme®) iki haftada bir intravenöz infüzyon olarak uygulanır. ERT ile ekstremite ağrısında azalma, böbrek fonksiyon kaybının yavaşlaması ve kalp hipertrofisinin ilerlemesinin baskılanması beklenir. Erken başlangıç, organ hasarının ilerlemesini baskılamak için önemlidir.
Migalastat (Galafold®) oral bir ilaçtır ve uygun (amenable) mutasyonlara sahip hastalarda endikedir. Değişime uğramış α-Gal A proteininin üç boyutlu yapısını stabilize ederek lizozoma taşınmasını kolaylaştırır.
Vorteks keratopatisi için spesifik bir tedavi gerekmez. Katarakt ilerleyip görme fonksiyonunu etkilerse katarakt cerrahisi endikedir.
QTedavi kornea semptomlarını iyileştirir mi?
A
Enzim replasman tedavisi (ERT) böbrek, kalp ve sinir semptomlarının ilerlemesini baskılayabilir, ancak korneadaki spiral şeklindeki birikintiler genellikle ERT ile düzelmez. Bununla birlikte, kornea bulguları görme üzerinde büyük bir etkiye sahip olmadığı için agresif tedavi gerekli değildir. Katarakt görmeyi etkiliyorsa cerrahi uygulanır.
6. Patofizyoloji ve ayrıntılı hastalık mekanizması
Fabry hastalığında, GLA gen mutasyonu nedeniyle α-Gal A aktivitesi azalır veya kaybolur. Sonuç olarak, bu enzim tarafından parçalanması gereken Gb3 ve onun deaçile formu olan Lyso-Gb3, lizozomda birikir. Biriken Gb3, hücresel işlev bozukluğuna, inflamatuar kaskadın aktivasyonuna ve oksidatif strese neden olarak ilerleyici çoklu organ hasarına yol açar.
Korneada, Gb3 limbustaki bazal kök hücrelerde birikir. Bu hücreler santrifüj yönde hareket ederken spiral birikinti paterni (cornea verticillata) oluşturur. Birikintiler kornea epitel bazal membran seviyesinde meydana gelir.
Lens epitel hücrelerinde ve kortikal liflerde Gb3 birikimi, arka subkapsüler alanda tekerlek teli benzeri (spoke-like) opasiteler oluşturur. Bu bulgu Fabry kataraktı olarak adlandırılır.
Vasküler endotel hücreleri ve düz kas hücrelerinde Gb3 birikimi, konjonktiva ve retinada damar kıvrımlanmasına ve genişlemesine neden olur. Retinada bilateral ana ven kıvrımlanması olarak gözlenir. Sistemik olarak, renal glomerül podositlerinde ve miyokard hücrelerinde birikim böbrek hasarına ve kardiyak hipertrofiye yol açar.
Fabry hastalığı tedavisinde geleneksel ERT’ye ek olarak çeşitli yeni tedavi yaklaşımları araştırılmaktadır.
Gen tedavisi, GLA geninin adeno-ilişkili virüs (AAV) vektörleri ile aktarılarak endojen α-Gal A üretimini sağlamayı amaçlayan köklü bir tedavi yöntemi olarak umut vaat etmektedir.
Substrat sentezi baskılama tedavisi (SRT), Gb3 sentezini baskılayan bir yaklaşımdır ve ERT’den farklı olarak intravenöz uygulama gerektirmeyen oral bir ilaç olarak geliştirilmektedir.
Yenidoğan taramasının genişletilmesi de önemli bir konudur. Erken tanı ile erken tedavi müdahalesi uzun dönem prognozu iyileştirebilir3).
Fenotipik çeşitlilik hakkında bilgi birikimi de artmaktadır. Aynı GLA mutasyonuna rağmen klasik tipten organa sınırlı tipe kadar farklı klinik tablolar görülebileceği1) ve kadın taşıyıcılarda X kromozomu inaktivasyon paterninin fenotipi öngörmediği bildirilmiştir. Klasik olmayan Fabry hastalığının nörolojik semptomları ile başvuran olgu raporlarında, beyaz cevher lezyonlarının incelenmesi sonucu Fabry hastalığı tanısına ulaşılan örnekler de vardır2).
Furia A, Ditaranto R, Biagini E, et al. Fabry disease in W162C mutation: a case report of two patients and a review of literature. BMC Neurol. 2024;24:113. PMCID: PMC10996216. doi:10.1186/s12883-024-03540-3.
Ferreira Tátá C, Massas M, Pinto F, Caçador N, Silva AL. Fabry Disease: A Atypical Presentation. Cureus. 2021;13(10):e18708. doi:10.7759/cureus.18708. PMID:34790463; PMCID:PMC8582620.
Starcea IM, Bodescu Amancei Ionescu L, Lazaruc TI, Lupu VV, Bogos RA, Ioniuc I, et al. Palm-Plant Pain, Sign of a Severe Systemic Disease? Case Report and Review of Literature. Genes. 2023;14(2). doi:10.3390/genes14020516. PMID:36833443; PMCID:PMC9957027.
Yanfang W, Juanjuan H, Shengli Z, Lei Y, Fei G. Case report and literature review: Fabry disease misdiagnosing as polymyalgia rheumatica. Medicine. 2023;102(44):e34630. doi:10.1097/MD.0000000000034630. PMID:37933054; PMCID:PMC10627660.
Makale metnini kopyalayıp tercih ettiğiniz yapay zeka asistanına yapıştırabilirsiniz.
Makale panoya kopyalandı
Aşağıdaki yapay zeka asistanlarından birini açın ve kopyalanan metni sohbet kutusuna yapıştırın.