Fabry disease is an inherited metabolic disorder caused by a deficiency of the lysosomal hydrolase α-galactosidase A (α-Gal A), leading to accumulation of glycosphingolipids, including globotriaosylceramide (Gb3), in cells throughout the body.
It exhibits X-linked recessive inheritance due to mutations in the GLA gene (Xq22.1). It was independently reported by Anderson and Fabry in 1898 2). The prevalence is estimated at about 1 in 10,000, but newborn screening reports higher frequencies (1:3,000 to 1:7,800) 3).
Clinical manifestations are broadly divided into classic and late-onset (non-classic) types. In the classic type, α-Gal A activity is almost absent, and multi-organ damage appears from early childhood. In the late-onset type, residual enzyme activity is present, and it may present as single-organ involvement of the heart or kidneys in adulthood 1, 2).
Disease type
Age of onset
Main affected organs
Classic type
Infancy to school age
Multiple organs (systemic)
Late-onset type
Adulthood
Heart and kidneys
QIs Fabry disease hereditary?
A
Yes, it is an X-linked recessive disorder. All daughters of a male patient are carriers, while sons are not affected. Female carriers may present a wide range of symptoms, from asymptomatic to severe, depending on the X-chromosome inactivation pattern. If you have a family member with this disease, genetic counseling is recommended.
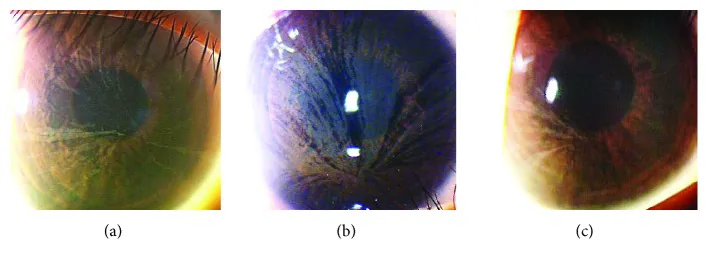
Yasuhito Ikegawa, Atsushi Shiraishi, Yasuhito Hayashi, Akiyoshi Ogimoto, et al. In Vivo Confocal Microscopic Observations of Vortex Keratopathy in Patients with Amiodarone-Induced Keratopathy and Fabry Disease 2018 Mar 21 J Ophthalmol. 2018 Mar 21; 2018:5315137 Figure 4. PMCID: PMC5884153. License: CC BY.
Slit-lamp biomicroscopy findings in three patients with Fabry disease. (a) Findings in a 32-year-old mother. (b) Findings in an 8-year-old daughter. (c) Findings in a 4-year-old daughter.
Visual impairment due to corneal verticillata is rare. In many cases, subjective complaints related to ocular symptoms are minimal.
Systemic symptoms include acroparesthesia (burning pain in the extremities) as the earliest symptom. Decreased sweating (hypohidrosis) and gastrointestinal symptoms also appear in childhood.
Location: At the level of the corneal epithelial basement membrane
Findings: Grayish, faint pigmentation spreads in a whorl-like pattern across the entire cornea
Carriers: Also frequently observed in female heterozygous carriers
Fabry Cataract
Location: Posterior subcapsular and lens cortex
Findings: Characteristic spoke-like opacities
Effect on vision: Usually mild, but advanced cases may require cataract surgery
Vascular Changes
Conjunctival vessels: Tortuosity and dilation observed
Retinal vessels: Characteristic bilateral tortuosity of the main retinal veins
Microaneurysms: May be observed in the conjunctiva and retina
In late-onset or cardiac-limited forms, typical corneal and skin findings may be absent. In a case of cardiac Fabry disease with the W162C mutation, ophthalmic evaluation revealed neither cornea verticillata nor angiokeratomas 1).
QDoes the corneal whorl pattern affect vision?
A
Cornea verticillata is a deposition in the superficial layer of the corneal epithelium and usually does not affect vision. It is often first detected by slit-lamp microscopy and is rarely noticed by the patient. However, if cataracts progress, vision loss may occur.
Fabry disease is caused by mutations in the GLA gene. Over 1000 types of mutations have been reported, and the clinical presentation varies greatly depending on the type of mutation 2).
Because it is an X-linked recessive disorder, males (hemizygotes) tend to have severe symptoms. Female carriers (heterozygotes) also exhibit a wide range of clinical presentations, from asymptomatic to severe classic forms, due to skewed X-chromosome inactivation 1). It is estimated that about 1% of female carriers develop symptoms, and corneal opacities are useful for screening in both sexes.
The main risk factor is a family history of Fabry disease. Patients with unexplained acroparesthesia, young-onset stroke, unexplained cardiac hypertrophy, or unexplained kidney disease should have this disease considered in the differential diagnosis 2, 4).
Slit-lamp microscopy is the most important examination. Vortex keratopathy is a characteristic finding and provides a clue to suspect this disease. The presence of cataract (posterior subcapsular spoke-like opacity) should also be confirmed. In non-classic cases, ocular findings may be absent 1); therefore, the absence of findings does not rule out this disease.
In males, a definitive diagnosis can be made by measuring α-Gal A enzyme activity. In female carriers, enzyme activity may be within the normal range due to X-inactivation, so genetic analysis is necessary 2). Plasma Lyso-Gb3 is a biomarker with high diagnostic sensitivity and is also useful for screening women 4).
The most important differential diagnosis for vortex keratopathy is drug-induced corneal deposition caused by amiodarone (an antiarrhythmic drug). The opacities resulting from amiodarone use closely resemble the vortex keratopathy of Fabry disease. Confirming the medication history is key to differentiation.
In addition, differentiation from drug-induced corneal deposits caused by chloroquine, indomethacin, and other drugs, as well as from other diseases that cause cornea verticillata, is necessary.
The fundamental treatment for Fabry disease is enzyme replacement therapy (ERT). Agalsidase alfa (Replagal®) or agalsidase beta (Fabrazyme®) is administered intravenously every two weeks. ERT is expected to reduce acroparesthesia, slow the decline of renal function, and suppress the progression of cardiac hypertrophy. Early initiation is important to suppress the progression of organ damage.
Migalastat (Galafold®) is an oral medication indicated for patients with amenable mutations. It stabilizes the conformation of the mutated α-Gal A protein and facilitates its transport to lysosomes.
Specific treatment for vortex keratopathy is not required. If cataracts progress and affect visual function, cataract surgery may be indicated.
QDoes treatment improve corneal symptoms?
A
Enzyme replacement therapy (ERT) can slow the progression of kidney, heart, and neurological symptoms, but it usually does not improve the corneal whorl-like deposits. However, since corneal findings do not significantly affect vision, active treatment is not necessary. Surgery is performed if cataracts affect vision.
6. Pathophysiology and Detailed Mechanism of Onset
In Fabry disease, mutations in the GLA gene reduce or eliminate the activity of α-Gal A. As a result, Gb3 and its deacylated form, Lyso-Gb3, which this enzyme normally breaks down, accumulate in lysosomes. Accumulated Gb3 causes cellular dysfunction, activation of inflammatory cascades, and oxidative stress, leading to progressive multi-organ damage.
In the cornea, Gb3 accumulates in the basal stem cells of the limbus. As these cells migrate centrifugally, they form a whorl-like deposition pattern (cornea verticillata). The deposits occur at the level of the corneal epithelial basement membrane.
In the lens, Gb3 accumulates in epithelial cells and cortical fibers, causing spoke-like opacities under the posterior capsule. This finding is called Fabry cataract.
Gb3 accumulation in vascular endothelial cells and smooth muscle cells causes tortuosity and dilation of conjunctival and retinal vessels. In the retina, it is observed as bilateral main venous tortuosity. Systemically, accumulation in podocytes of renal glomeruli and cardiomyocytes leads to kidney damage and cardiac hypertrophy.
In addition to conventional ERT, several novel therapeutic approaches for Fabry disease are being studied.
Gene therapy is expected as a curative treatment that introduces the GLA gene via adeno-associated virus (AAV) vectors to produce endogenous α-Gal A.
Substrate reduction therapy (SRT) is an approach that suppresses the synthesis of Gb3 itself, and unlike ERT, it is being developed as an oral drug that does not require intravenous administration.
Expansion of newborn screening is also an important issue. Early diagnosis and early treatment intervention may improve long-term prognosis3).
Knowledge about phenotypic diversity is also accumulating. It has been reported that even with the same GLA mutation, different clinical presentations ranging from classic to organ-limited forms can occur1), and that X-chromosome inactivation patterns in female carriers do not predict phenotype. In case reports of non-classic Fabry disease presenting with neurological symptoms, there are examples where detailed examination of white matter lesions led to the diagnosis of Fabry disease2).
Furia A, Ditaranto R, Biagini E, et al. Fabry disease in W162C mutation: a case report of two patients and a review of literature. BMC Neurol. 2024;24:113. PMCID: PMC10996216. doi:10.1186/s12883-024-03540-3.
Ferreira Tátá C, Massas M, Pinto F, Caçador N, Silva AL. Fabry Disease: A Atypical Presentation. Cureus. 2021;13(10):e18708. doi:10.7759/cureus.18708. PMID:34790463; PMCID:PMC8582620.
Starcea IM, Bodescu Amancei Ionescu L, Lazaruc TI, Lupu VV, Bogos RA, Ioniuc I, et al. Palm-Plant Pain, Sign of a Severe Systemic Disease? Case Report and Review of Literature. Genes. 2023;14(2). doi:10.3390/genes14020516. PMID:36833443; PMCID:PMC9957027.
Yanfang W, Juanjuan H, Shengli Z, Lei Y, Fei G. Case report and literature review: Fabry disease misdiagnosing as polymyalgia rheumatica. Medicine. 2023;102(44):e34630. doi:10.1097/MD.0000000000034630. PMID:37933054; PMCID:PMC10627660.
Copy the article text and paste it into your preferred AI assistant.
Article copied to clipboard
Open an AI assistant below and paste the copied text into the chat box.