La malattia di Fabry è una malattia metabolica ereditaria causata da un deficit di α-galattosidasi A (α-Gal A), un enzima lisosomiale, che porta all’accumulo di glicosfingolipidi, tra cui il globotriaosilceramide (Gb3), nelle cellule di tutto il corpo.
Presenta ereditarietà recessiva legata all’X dovuta a mutazioni nel gene GLA (Xq22.1). Fu descritta indipendentemente da Anderson e Fabry nel 1898 2). La prevalenza è stimata in circa 1 persona su 10.000, ma lo screening neonatale riporta una frequenza più alta (1:3.000–1:7.800) 3).
Il quadro clinico si divide in forma classica e forma tardiva (non classica). Nella forma classica, l’attività dell’α-Gal A è quasi assente e fin dall’infanzia compaiono danni multiorgano. Nella forma tardiva, vi è un’attività enzimatica residua e la malattia può manifestarsi in età adulta come danno d’organo singolo (cuore o rene) 1, 2).
Tipo di malattia
Età di insorgenza
Principali organi colpiti
Classico
Prima infanzia – età scolare
Multi-organo (sistemico)
Forma tardiva
Età adulta
Cuore e reni
QLa malattia di Fabry è ereditaria?
A
Sì, è una malattia recessiva legata al cromosoma X. Tutte le figlie di un paziente maschio sono portatrici, mentre i figli maschi non ereditano la malattia. Le donne portatrici possono presentare un’ampia gamma di sintomi, da asintomatici a gravi, a seconda del pattern di inattivazione del cromosoma X. Se in famiglia c’è un caso di questa malattia, si consiglia di sottoporsi a consulenza genetica.
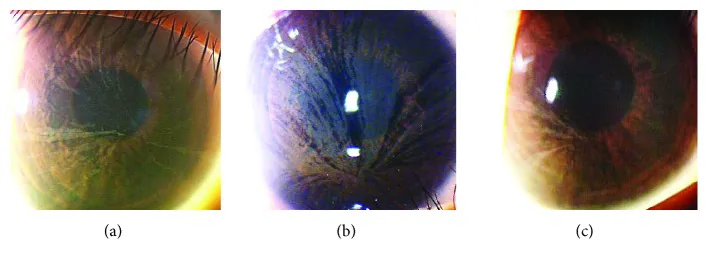
Yasuhito Ikegawa, Atsushi Shiraishi, Yasuhito Hayashi, Akiyoshi Ogimoto, et al. In Vivo Confocal Microscopic Observations of Vortex Keratopathy in Patients with Amiodarone-Induced Keratopathy and Fabry Disease 2018 Mar 21 J Ophthalmol. 2018 Mar 21; 2018:5315137 Figure 4. PMCID: PMC5884153. License: CC BY.
Risultati dell’esame con lampada a fessura di tre pazienti con malattia di Fabry. (a) Risultati della madre di 32 anni. (b) Risultati della figlia di 8 anni. (c) Risultati della figlia di 4 anni.
La riduzione dell’acuità visiva dovuta all’opacità corneale a spirale è rara. Spesso, i sintomi oculari causano poche lamentele soggettive.
I sintomi sistemici includono dolore urente alle estremità (acroparestesia) come sintomo più precoce. Anche la ridotta sudorazione (ipoidrosi) e i sintomi gastrointestinali compaiono fin dall’infanzia.
Età di insorgenza: osservabile a partire da circa 6 anni
Sede: a livello della membrana basale dell’epitelio corneale
Reperto: depositi pigmentati grigiastri pallidi che si diffondono a vortice su tutta la cornea
Portatori: osservato frequentemente anche nelle donne eterozigoti portatrici
Cataratta di Fabry
Sede : sottocapsulare posteriore e corteccia del cristallino
Reperto : opacità a forma di raggi di ruota (spoke-like) caratteristiche
Impatto sulla vista : di solito lieve, ma nei casi avanzati può essere indicato l’intervento di cataratta
Alterazioni vascolari
Vasi congiuntivali : tortuosità e dilatazione
Vasi retinici: tortuosità bilaterale delle vene principali, caratteristica
Microaneurismi: possono essere osservati nella congiuntiva e nella retina
Nelle forme tardive o cardiache, i tipici segni corneali e cutanei possono essere assenti. In un caso di malattia di Fabry cardiaca con mutazione W162C, la valutazione oftalmica non ha mostrato né cornea verticillata né angiocheratomi1).
QI vortici corneali influenzano la vista?
A
La cornea verticillata è un deposito negli strati superficiali dell’epitelio corneale e di solito non influisce sulla vista. Viene spesso scoperta per la prima volta all’esame con lampada a fessura, e il paziente stesso quasi mai se ne accorge. Tuttavia, in caso di progressione della cataratta, può verificarsi una riduzione dell’acuità visiva.
La causa della malattia di Fabry è una mutazione del gene GLA. Sono state riportate oltre 1000 mutazioni diverse e il quadro clinico varia notevolmente a seconda del tipo di mutazione2).
Essendo una malattia recessiva legata all’X, i maschi (emizigoti) sono più soggetti a forme gravi. Anche le donne portatrici (eterozigoti) presentano un quadro clinico variabile, da asintomatico a grave simile alla forma classica, a causa di un’inattivazione sbilanciata del cromosoma X1). Circa l’1% delle donne portatrici sviluppa sintomi, e la conferma di opacità corneale è utile per lo screening in entrambi i sessi.
Il principale fattore di rischio è una storia familiare di malattia di Fabry. In pazienti con dolore acrale inspiegato, ictus giovanile, cardiomegalia inspiegata o insufficienza renale inspiegata, questa malattia deve essere considerata nella diagnosi differenziale2, 4).
L’esame con lampada a fessura è il più importante. La cornea verticillata è un reperto caratteristico e costituisce un’opportunità per sospettare questa malattia. Verificare anche la presenza di cataratta (opacità subcapsulare posteriore a forma di raggio). Nei casi non classici, i reperti oftalmologici possono essere assenti 1); l’assenza di reperti non deve escludere questa malattia.
Negli uomini, la diagnosi definitiva è possibile mediante la misurazione dell’attività dell’α-Gal A. Nelle donne portatrici, a causa dell’inattivazione del cromosoma X, l’attività enzimatica può rientrare nell’intervallo normale, pertanto è necessaria l’analisi genetica 2). Il Lyso-Gb3 plasmatico è un biomarcatore con elevata sensibilità diagnostica, utile anche per lo screening nelle donne 4).
La diagnosi differenziale più importante della cornea verticillata è la deposizione corneale farmaco-indotta da amiodarone (farmaco antiaritmico). Le opacità causate dall’assunzione di amiodarone sono molto simili alla cornea verticillata della malattia di Fabry. La verifica della storia farmacologica è la chiave per la diagnosi differenziale.
Inoltre, è necessaria la differenziazione da altre deposizioni corneali farmaco-indotte come quelle da clorochina, indometacina, e da altre malattie che causano cornea verticillata.
Il trattamento di base della malattia di Fabry è la terapia enzimatica sostitutiva (enzyme replacement therapy: ERT). L’agalsidasi alfa (Replagal®) o l’agalsidasi beta (Fabrazyme®) vengono somministrate per infusione endovenosa ogni due settimane. L’ERT può ridurre il dolore alle estremità, rallentare il declino della funzione renale e inibire la progressione dell’ipertrofia cardiaca. L’inizio precoce è importante per rallentare la progressione del danno d’organo.
Il migalastat (Galafold®) è un farmaco orale indicato per i pazienti con mutazioni amendabili. Stabilizza la struttura tridimensionale della proteina α-Gal A denaturata e ne favorisce il trasporto verso il lisosoma.
Non è necessario alcun trattamento specifico per la cornea verticillata. In caso di cataratta progressiva che compromette la funzione visiva, è indicato l’intervento di cataratta.
QIl trattamento migliora i sintomi corneali?
A
La terapia enzimatica sostitutiva (ERT) può rallentare la progressione dei sintomi renali, cardiaci e neurologici, ma di solito non migliora i depositi corneali a vortice. Tuttavia, poiché questi reperti corneali non influenzano significativamente la vista, non è necessario un trattamento aggressivo. Se la cataratta compromette la vista, si procede con un intervento chirurgico.
Nella malattia di Fabry, le mutazioni del gene GLA causano una riduzione o perdita dell’attività dell’α-Gal A. Di conseguenza, Gb3 e la sua forma deacilata Lyso-Gb3, che normalmente dovrebbero essere degradati da questo enzima, si accumulano nei lisosomi. L’accumulo di Gb3 provoca disfunzione cellulare, attivazione di cascate infiammatorie e stress ossidativo, portando a un danno multiorgano progressivo.
Nella cornea, Gb3 si accumula nelle cellule staminali basali del limbo. Durante la migrazione centrifuga di queste cellule, si forma un pattern di depositi a vortice (cornea verticillata). I depositi si verificano a livello della membrana basale dell’epitelio corneale.
Nel cristallino, il Gb3 si accumula nelle cellule epiteliali e nelle fibre corticali, causando un’opacità a forma di raggi (spoke-like) sotto la capsula posteriore. Questa è chiamata cataratta di Fabry.
L’accumulo di Gb3 nelle cellule endoteliali vascolari e nelle cellule muscolari lisce provoca tortuosità e dilatazione dei vasi congiuntivali e retinici. Nella retina, si osserva come tortuosità bilaterale delle vene principali. A livello sistemico, l’accumulo nei podociti glomerulari renali e nei cardiomiociti causa danno renale e ipertrofia cardiaca.
Oltre alla ERT convenzionale, diversi nuovi approcci terapeutici sono in fase di studio per la malattia di Fabry.
La terapia genica, che introduce il gene GLA tramite un vettore virale adeno-associato (AAV) per produrre α-Gal A in modo endogeno, è considerata un trattamento curativo promettente.
La terapia di riduzione del substrato (SRT) è un approccio che inibisce la sintesi stessa del Gb3. A differenza dell’ERT, viene sviluppata come farmaco orale che non richiede somministrazione endovenosa.
L’espansione dello screening neonatale è anche una questione importante. Un intervento terapeutico precoce grazie a una diagnosi precoce potrebbe migliorare la prognosi a lungo termine3).
Si stanno accumulando anche conoscenze sulla diversità fenotipica. È stato riportato che, anche con la stessa mutazione GLA, possono presentarsi quadri clinici diversi, dalla forma classica a quella limitata a un organo1) e che il pattern di inattivazione del cromosoma X nelle donne portatrici non predice il fenotipo. In casi clinici di malattia di Fabry non classica con sintomi neurologici come principale lamentela, ci sono esempi in cui la diagnosi di malattia di Fabry è stata raggiunta dopo un esame approfondito delle lesioni della sostanza bianca cerebrale2).
Furia A, Ditaranto R, Biagini E, et al. Fabry disease in W162C mutation: a case report of two patients and a review of literature. BMC Neurol. 2024;24:113. PMCID: PMC10996216. doi:10.1186/s12883-024-03540-3.
Ferreira Tátá C, Massas M, Pinto F, Caçador N, Silva AL. Fabry Disease: A Atypical Presentation. Cureus. 2021;13(10):e18708. doi:10.7759/cureus.18708. PMID:34790463; PMCID:PMC8582620.
Starcea IM, Bodescu Amancei Ionescu L, Lazaruc TI, Lupu VV, Bogos RA, Ioniuc I, et al. Palm-Plant Pain, Sign of a Severe Systemic Disease? Case Report and Review of Literature. Genes. 2023;14(2). doi:10.3390/genes14020516. PMID:36833443; PMCID:PMC9957027.
Yanfang W, Juanjuan H, Shengli Z, Lei Y, Fei G. Case report and literature review: Fabry disease misdiagnosing as polymyalgia rheumatica. Medicine. 2023;102(44):e34630. doi:10.1097/MD.0000000000034630. PMID:37933054; PMCID:PMC10627660.
Copia il testo dell'articolo e incollalo nell'assistente IA che preferisci.
Articolo copiato negli appunti
Apri un assistente IA qui sotto e incolla il testo copiato nella chat.