بیماری فابری یک بیماری متابولیک ارثی است که در اثر کمبود آنزیم لیزوزومی α-گالاکتوزیداز A (α-Gal A) ایجاد میشود و منجر به تجمع گلیکوسفنگولیپیدها، به ویژه گلوبوتریائوسیل سرامید (Gb3)، در سلولهای سراسر بدن میگردد.
این بیماری با الگوی وراثت وابسته به X مغلوب ناشی از جهش در ژن GLA (Xq22.1) تظاهر میکند. اولین بار در سال ۱۸۹۸ توسط اندرسون و فابری به طور مستقل گزارش شد 2). شیوع آن حدود ۱ در ۱۰٬۰۰۰ نفر تخمین زده میشود، اما در غربالگری نوزادان فراوانی بالاتر (۱:۳٬۰۰۰ تا ۱:۷٬۸۰۰) نیز گزارش شده است 3).
تصویر بالینی به دو نوع کلاسیک و دیررس (غیرکلاسیک) تقسیم میشود. در نوع کلاسیک، فعالیت α-Gal A تقریباً از بین رفته و از دوران کودکی آسیب چندعضوی ظاهر میشود. در نوع دیررس، فعالیت آنزیمی باقیمانده وجود دارد و ممکن است در بزرگسالی به صورت آسیب تکعضوی قلبی یا کلیوی بروز کند 1, 2).
نوع بیماری
زمان شروع
ارگانهای اصلی درگیر
کلاسیک
نوزادی تا سن مدرسه
چندارگانی (کل بدن)
نوع دیررس
بزرگسالی
قلب و کلیه
Qآیا بیماری فابری ارثی است؟
A
بله، این بیماری با وراثت وابسته به X مغلوب منتقل میشود. همه دختران یک بیمار مرد ناقل میشوند، اما پسران به ارث نمیبرند. زنان ناقل بسته به الگوی غیرفعالسازی کروموزوم X ممکن است طیف وسیعی از علائم از بدون علامت تا شدید را نشان دهند. اگر در خانواده شما فردی مبتلا به این بیماری وجود دارد، توصیه میشود مشاوره ژنتیک دریافت کنید.
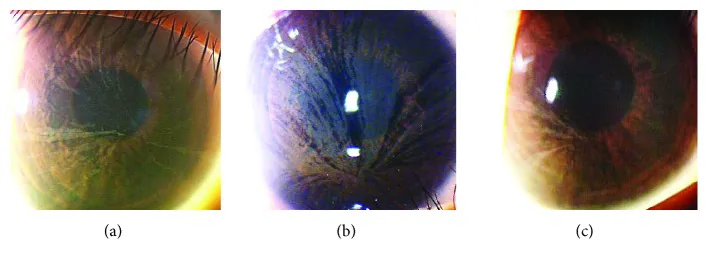
Yasuhito Ikegawa, Atsushi Shiraishi, Yasuhito Hayashi, Akiyoshi Ogimoto, et al. In Vivo Confocal Microscopic Observations of Vortex Keratopathy in Patients with Amiodarone-Induced Keratopathy and Fabry Disease 2018 Mar 21 J Ophthalmol. 2018 Mar 21; 2018:5315137 Figure 4. PMCID: PMC5884153. License: CC BY.
یافتههای معاینه با میکروسکوپ اسلیت لمپ در سه بیمار مبتلا به بیماری فابری. (a) مادر ۳۲ ساله. (b) دختر ۸ ساله. (c) دختر ۴ ساله.
یافته: رسوبات رنگدانهای خاکستری کمرنگ که به صورت حلزونی در سراسر قرنیه گسترش مییابند
ناقلین: در زنان ناقل هتروزیگوت نیز با فراوانی بالا مشاهده میشود
آبمروارید فابری
محل: زیر کپسول خلفی و قشر عدسی
یافته: کدورتهای شعاعی (spoke-like) مشخصه
تأثیر بر بینایی: معمولاً خفیف است اما در موارد پیشرفته نیاز به جراحی آبمروارید دارد
تغییرات عروقی
عروق ملتحمه: پیچخوردگی و گشادگی مشاهده میشود
رگهای شبکیه: پیچخوردگی وریدهای اصلی دوطرفه مشخصه است
آنوریسمهای میکروسکوپی: ممکن است در ملتحمه و شبکیه دیده شوند
در نوع دیررس یا محدود به قلب، ممکن است یافتههای معمول قرنیه و پوست وجود نداشته باشند. در یک مورد از بیماری فابری قلبی با جهش W162C، گزارش شده است که در ارزیابی چشمی نه قرنیه حلزونی و نه آنژیوکراتوما مشاهده نشد1).
Qآیا الگوی مارپیچی قرنیه بر بینایی تأثیر میگذارد؟
A
قرنیه حلزونی (cornea verticillata) رسوبی در لایه سطحی اپیتلیوم قرنیه است که معمولاً بر بینایی تأثیر نمیگذارد. اغلب برای اولین بار در معاینه با لامپ شکاف دار کشف میشود و خود بیمار به ندرت متوجه آن میشود. با این حال، اگر آب مروارید پیشرفت کند، ممکن است کاهش بینایی رخ دهد.
علت بیماری فابری جهش در ژن GLA است. بیش از ۱۰۰۰ نوع جهش گزارش شده است و تصویر بالینی بسته به نوع جهش بسیار متفاوت است 2).
از آنجایی که این بیماری به صورت وابسته به X مغلوب به ارث میرسد، مردان (همیزیگوت) بیشتر در معرض ابتلا به نوع شدید هستند. زنان ناقل (هتروزیگوت) نیز به دلیل عدم تعادل در غیرفعالسازی کروموزوم X، طیف بالینی متنوعی از بدون علامت تا نوع شدید کلاسیک را نشان میدهند 1). حدود ۱٪ از زنان ناقل نیز علائم را بروز میدهند و تشخیص کدورت قرنیه برای غربالگری در هر دو جنس مفید است.
عامل خطر اصلی سابقه خانوادگی بیماری فابری است. در بیمارانی که درد انتهای اندامها با علت ناشناخته، سکته مغزی در سنین پایین، بزرگشدگی قلب با علت ناشناخته، یا نارسایی کلیوی با علت ناشناخته دارند، باید این بیماری را در تشخیص افتراقی در نظر گرفت 2, 4).
معاینه با لامپ شکاف مهمترین است. قرنیه حلزونی یک یافته مشخص است و باعث شک به این بیماری میشود. وجود آب مروارید (کدورت محوری زیرکپسول خلفی) نیز بررسی میشود. در موارد غیرکلاسیک، ممکن است یافتههای چشمی وجود نداشته باشند1)، بنابراین عدم وجود یافته نباید این بیماری را رد کند.
در مردان، تشخیص قطعی با اندازهگیری فعالیت آنزیم α-Gal A امکانپذیر است. در زنان ناقل، به دلیل تأثیر غیرفعالسازی کروموزوم X، فعالیت آنزیم ممکن است در محدوده طبیعی باشد، بنابراین تجزیه و تحلیل ژنتیکی ضروری است2). Lyso-Gb3 پلاسما یک بیومارکر با حساسیت تشخیصی بالا است که برای غربالگری زنان نیز مفید میباشد4).
مهمترین تشخیص افتراقی برای قرنیه حلزونی شکل، رسوبات قرنیهای ناشی از داروی آمیودارون (داروی ضد آریتمی) است. کدورتهای ناشی از مصرف آمیودارون ظاهری بسیار شبیه به قرنیه حلزونی شکل در بیماری فابری دارند. بررسی سابقه مصرف دارو کلید تشخیص افتراقی است.
علاوه بر این، تشخیص افتراقی از رسوبات قرنیهای ناشی از داروهایی مانند کلروکین و ایندومتاسین و همچنین سایر بیماریهای ایجادکننده cornea verticillata ضروری است.
درمان اساسی بیماری فابری درمان جایگزینی آنزیم (enzyme replacement therapy: ERT) است. آگالسیداز آلفا (ریپلگال®) یا آگالسیداز بتا (فابرازایم®) هر دو هفته یک بار به صورت تزریق وریدی تجویز میشود. ERT میتواند باعث کاهش درد انتهای اندامها، کند شدن کاهش عملکرد کلیه و مهار پیشرفت هیپرتروفی قلبی شود. شروع زودهنگام برای مهار پیشرفت آسیب اندامها مهم است.
میگالاستات (گالافولد®) یک داروی خوراکی است که برای بیماران دارای جهشهای قابل درمان (amenable) تجویز میشود. این دارو ساختار سهبعدی پروتئین α-Gal A تغییر یافته را تثبیت کرده و انتقال آن به لیزوزوم را تسهیل میکند.
برای کدورت قرنیه به شکل گردابی درمان خاصی لازم نیست. اگر آب مروارید پیشرفت کرده و بر عملکرد بینایی تأثیر بگذارد، جراحی آب مروارید اندیکاسیون دارد.
Qآیا درمان علائم قرنیه را بهبود میبخشد؟
A
درمان جایگزینی آنزیمی (ERT) میتواند پیشرفت علائم کلیوی، قلبی و عصبی را مهار کند، اما رسوبات مارپیچی قرنیه معمولاً با ERT بهبود نمییابند. با این حال، از آنجایی که یافتههای قرنیه تأثیر عمدهای بر بینایی ندارند، درمان تهاجمی ضروری نیست. اگر آب مروارید بر بینایی تأثیر بگذارد، جراحی انجام میشود.
در بیماری فابری، جهش در ژن GLA باعث کاهش یا فقدان فعالیت α-Gal A میشود. در نتیجه، Gb3 و شکل دِآسیله آن یعنی Lyso-Gb3 که باید توسط این آنزیم تجزیه شوند، در لیزوزوم تجمع مییابند. Gb3 تجمعیافته باعث اختلال عملکرد سلولی، فعالسازی آبشار التهابی و استرس اکسیداتیو میشود و منجر به آسیب پیشرونده چندعضوی میگردد.
در قرنیه، Gb3 در سلولهای بنیادی پایه لیمبوس تجمع مییابد. این سلولها در حین حرکت گریز از مرکز، الگوی رسوب مارپیچی (cornea verticillata) را تشکیل میدهند. رسوبات در سطح غشای پایه اپیتلیوم قرنیه رخ میدهند.
در عدسی، Gb3 در سلولهای اپیتلیال و الیاف قشری تجمع یافته و کدورتهای شعاعیشکل (spoke-like) در زیر کپسول خلفی ایجاد میکند. این یافته آب مروارید فابری نامیده میشود.
تجمع Gb3 در سلولهای اندوتلیال عروقی و سلولهای عضله صاف باعث پیچخوردگی و گشادشدگی عروق ملتحمه و شبکیه میشود. در شبکیه، به صورت پیچخوردگی دوطرفه وریدهای اصلی مشاهده میشود. به طور سیستمیک، تجمع در پودوسیتهای گلومرول کلیه و سلولهای میوکارد باعث آسیب کلیوی و هیپرتروفی قلبی میشود.
علاوه بر ERT سنتی، چندین رویکرد درمانی جدید برای بیماری فابری در حال بررسی است.
ژن درمانی با وارد کردن ژن GLA از طریق ناقلهای ویروسی مرتبط با آدنو (AAV) برای تولید درونزاد α-Gal A به عنوان یک درمان ریشهای امیدوارکننده است.
درمان مهار سنتز بستر (SRT) رویکردی است که سنتز Gb3 را مهار میکند و برخلاف ERT، به صورت داروی خوراکی بدون نیاز به تزریق وریدی در حال توسعه است.
غربالگری نوزادان نیز یک موضوع مهم است. مداخله زودهنگام با تشخیص زودهنگام ممکن است پیشآگهی طولانیمدت را بهبود بخشد3).
دانش در مورد تنوع فنوتیپی نیز در حال انباشته شدن است. گزارش شده است که حتی با یک جهش GLA یکسان، تظاهرات بالینی از نوع کلاسیک تا محدود به اندام متفاوت است1) و الگوی غیرفعالسازی کروموزوم X در زنان ناقل، فنوتیپ را پیشبینی نمیکند. در گزارشهای موردی با شکایت اصلی علائم عصبی بیماری فابری غیرکلاسیک، مواردی وجود دارد که از بررسی ضایعات ماده سفید مغز به تشخیص بیماری فابری منجر شده است2).
Furia A, Ditaranto R, Biagini E, et al. Fabry disease in W162C mutation: a case report of two patients and a review of literature. BMC Neurol. 2024;24:113. PMCID: PMC10996216. doi:10.1186/s12883-024-03540-3.
Ferreira Tátá C, Massas M, Pinto F, Caçador N, Silva AL. Fabry Disease: A Atypical Presentation. Cureus. 2021;13(10):e18708. doi:10.7759/cureus.18708. PMID:34790463; PMCID:PMC8582620.
Starcea IM, Bodescu Amancei Ionescu L, Lazaruc TI, Lupu VV, Bogos RA, Ioniuc I, et al. Palm-Plant Pain, Sign of a Severe Systemic Disease? Case Report and Review of Literature. Genes. 2023;14(2). doi:10.3390/genes14020516. PMID:36833443; PMCID:PMC9957027.
Yanfang W, Juanjuan H, Shengli Z, Lei Y, Fei G. Case report and literature review: Fabry disease misdiagnosing as polymyalgia rheumatica. Medicine. 2023;102(44):e34630. doi:10.1097/MD.0000000000034630. PMID:37933054; PMCID:PMC10627660.
متن مقاله را کپی کنید و در دستیار هوش مصنوعی دلخواه خود بچسبانید.
مقاله در کلیپبورد کپی شد
یکی از دستیارهای هوش مصنوعی زیر را باز کنید و متن کپیشده را در کادر گفتگو بچسبانید.