La maladie de Fabry est une maladie métabolique héréditaire causée par un déficit en α-galactosidase A (α-Gal A), une enzyme lysosomale, entraînant l’accumulation de glycosphingolipides, notamment le globotriaosylcéramide (Gb3), dans les cellules de tout l’organisme.
Elle est liée à l’X et due à des mutations du gène GLA (Xq22.1). Elle a été décrite indépendamment par Anderson et Fabry en 1898 2). La prévalence est estimée à environ 1 personne sur 10 000, mais le dépistage néonatal rapporte une fréquence plus élevée (1:3 000 à 1:7 800) 3).
Les manifestations cliniques sont divisées en forme classique et forme tardive (non classique). Dans la forme classique, l’activité de l’α-Gal A est presque absente, et des lésions multi-organes apparaissent dès l’enfance. Dans la forme tardive, une activité enzymatique résiduelle persiste et la maladie peut se manifester à l’âge adulte par une atteinte cardiaque ou rénale isolée 1, 2).
Type de maladie
Âge d’apparition
Principaux organes touchés
Classique
Petite enfance à âge scolaire
Multi-organes (systémique)
Forme tardive
Âge adulte
Cœur et reins
QLa maladie de Fabry est-elle héréditaire ?
A
Oui, c’est une maladie liée à l’X récessive. Toutes les filles d’un patient masculin sont porteuses, mais les fils ne sont pas affectés. Les femmes porteuses peuvent présenter une large gamme de symptômes, allant d’asymptomatiques à sévères, en fonction du motif d’inactivation du chromosome X. Si un membre de votre famille est atteint, il est recommandé de consulter un conseiller en génétique.
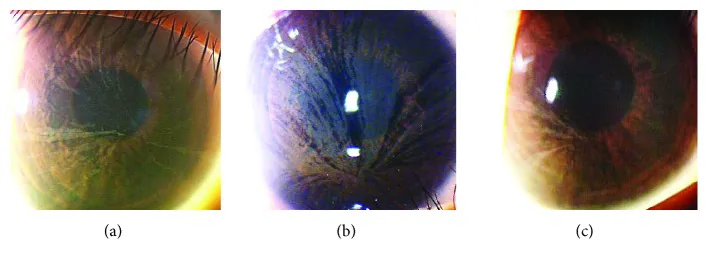
Yasuhito Ikegawa, Atsushi Shiraishi, Yasuhito Hayashi, Akiyoshi Ogimoto, et al. In Vivo Confocal Microscopic Observations of Vortex Keratopathy in Patients with Amiodarone-Induced Keratopathy and Fabry Disease 2018 Mar 21 J Ophthalmol. 2018 Mar 21; 2018:5315137 Figure 4. PMCID: PMC5884153. License: CC BY.
Observations à la lampe à fente de trois patients atteints de la maladie de Fabry. (a) Mère de 32 ans. (b) Fille de 8 ans. (c) Fille de 4 ans.
La baisse de l’acuité visuelle due à l’opacité cornéenne en spirale est rare. Dans la plupart des cas, les plaintes subjectives liées aux symptômes oculaires sont rares.
Les symptômes systémiques comprennent des douleurs brûlantes aux extrémités (acroparesthésie) comme symptôme le plus précoce. Une diminution de la transpiration (hypohidrose) et des symptômes gastro-intestinaux apparaissent également dès l’enfance.
Localisation : niveau de la membrane basale de l’épithélium cornéen
Aspect : dépôts pigmentaires grisâtres pâles s’étendant en tourbillon sur toute la cornée
Porteurs : également observé fréquemment chez les femmes hétérozygotes porteuses
Cataracte de Fabry
Localisation : sous-capsulaire postérieure et cortex du cristallin
Aspect : opacités en forme de rayons de roue (spoke-like)
Impact sur la vision : généralement mineur, mais peut nécessiter une chirurgie de la cataracte dans les cas avancés
Modifications vasculaires
Vaisseaux conjonctivaux : tortuosité et dilatation
Vaisseaux rétiniens : tortuosité caractéristique des veines principales, bilatérale
Microanévrismes : peuvent être observés dans la conjonctive et la rétine
Dans les formes tardives ou cardiaques, les signes typiques cornéens et cutanés peuvent être absents. Un cas de maladie de Fabry cardiaque avec mutation W162C n’a montré ni cornée verticillée ni angiokératomes à l’évaluation ophtalmologique1).
QLes tourbillons cornéens affectent-ils la vision ?
A
La cornée verticillée (cornea verticillata) est un dépôt dans les couches superficielles de l’épithélium cornéen et n’affecte généralement pas la vision. Elle est souvent découverte pour la première fois lors d’un examen à la lampe à fente, et le patient lui-même ne la remarque presque jamais. Cependant, une baisse de l’acuité visuelle peut survenir en cas de progression de la cataracte.
La cause de la maladie de Fabry est une mutation du gène GLA. Plus de 1000 mutations ont été rapportées, et le tableau clinique varie considérablement selon le type de mutation2).
Étant une maladie récessive liée à l’X, les hommes (hémizygotes) sont plus susceptibles de développer des formes graves. Les femmes conductrices (hétérozygotes) présentent également un tableau clinique varié, allant de l’absence de symptômes à une forme sévère proche de la forme classique, en raison d’une inactivation biaisée du chromosome X1). Environ 1% des femmes conductrices développent des symptômes, et la détection d’une opacité cornéenne est utile pour le dépistage chez les deux sexes.
Le principal facteur de risque est un antécédent familial de maladie de Fabry. Chez les patients présentant des douleurs acrales inexpliquées, un accident vasculaire cérébral juvénile, une hypertrophie cardiaque inexpliquée ou une insuffisance rénale inexpliquée, cette maladie doit être envisagée dans le diagnostic différentiel2, 4).
L’examen à la lampe à fente est le plus important. La cornée verticillée est une constatation caractéristique et constitue une occasion de suspecter cette maladie. Vérifiez également la présence de cataracte (opacité sous-capsulaire postérieure en rayon de roue). Dans les cas non classiques, les signes ophtalmologiques peuvent être absents 1) ; l’absence de signes ne doit pas exclure cette maladie.
Chez les hommes, le diagnostic définitif peut être posé par la mesure de l’activité de l’α-Gal A. Chez les femmes conductrices, en raison de l’inactivation du chromosome X, l’activité enzymatique peut se situer dans la plage normale, ce qui nécessite une analyse génétique 2). Le Lyso-Gb3 plasmatique est un biomarqueur à haute sensibilité diagnostique, également utile pour le dépistage chez les femmes 4).
Le diagnostic différentiel le plus important de la cornée verticillée est la dépôt cornéen médicamenteux dû à l’amiodarone (antiarythmique). Les opacités causées par la prise d’amiodarone ressemblent beaucoup à la cornée verticillée de la maladie de Fabry. La vérification des antécédents médicamenteux est essentielle pour le diagnostic différentiel.
En outre, il est nécessaire de différencier d’autres dépôts cornéens médicamenteux comme ceux dus à la chloroquine ou à l’indométacine, ainsi que d’autres maladies provoquant une cornée verticillée.
Le traitement de fond de la maladie de Fabry est le traitement enzymatique substitutif (enzyme replacement therapy: ERT). L’agalsidase alfa (Replagal®) ou l’agalsidase bêta (Fabrazyme®) sont administrés par perfusion intraveineuse toutes les deux semaines. L’ERT permet de réduire les douleurs des extrémités, de ralentir la dégradation de la fonction rénale et de freiner la progression de l’hypertrophie cardiaque. Un début précoce est important pour ralentir la progression des lésions organiques.
Le migalastat (Galafold®) est un médicament oral indiqué chez les patients présentant des mutations amendables. Il stabilise la conformation tridimensionnelle de la protéine α-Gal A dénaturée et favorise son transport vers le lysosome.
Aucun traitement spécifique n’est nécessaire pour la cornée verticillée. En cas de cataracte évolutive affectant la fonction visuelle, une chirurgie de la cataracte peut être indiquée.
QLe traitement améliore-t-il les symptômes cornéens ?
A
La thérapie enzymatique substitutive (ERT) peut ralentir la progression des symptômes rénaux, cardiaques et neurologiques, mais elle n’améliore généralement pas les dépôts cornéens en tourbillon. Cependant, comme ces dépôts n’affectent pas significativement l’acuité visuelle, un traitement agressif n’est pas nécessaire. Si la cataracte affecte la vision, une chirurgie peut être réalisée.
Dans la maladie de Fabry, les mutations du gène GLA entraînent une diminution ou une perte d’activité de l’α-Gal A. En conséquence, le Gb3 et son forme déacylée, le Lyso-Gb3, qui devraient normalement être dégradés par cette enzyme, s’accumulent dans les lysosomes. L’accumulation de Gb3 provoque un dysfonctionnement cellulaire, l’activation de cascades inflammatoires et un stress oxydatif, conduisant à une atteinte multi-organique progressive.
Dans la cornée, le Gb3 s’accumule dans les cellules souches basales du limbe. Lors de la migration centrifuge de ces cellules, elles forment un motif de dépôt en tourbillon (cornea verticillata). Les dépôts se produisent au niveau de la membrane basale de l’épithélium cornéen.
Dans le cristallin, le Gb3 s’accumule dans les cellules épithéliales et les fibres corticales, provoquant une opacité en forme de rayons (spoke-like) sous la capsule postérieure. C’est ce que l’on appelle la cataracte de Fabry.
L’accumulation de Gb3 dans les cellules endothéliales et les cellules musculaires lisses vasculaires provoque une tortuosité et une dilatation des vaisseaux conjonctivaux et rétiniens. Dans la rétine, elle est observée comme une tortuosité bilatérale des veines principales. Au niveau systémique, l’accumulation dans les podocytes glomérulaires rénaux et les cardiomyocytes entraîne une insuffisance rénale et une hypertrophie cardiaque.
En plus de l’ERT conventionnelle, plusieurs nouvelles approches thérapeutiques sont étudiées pour la maladie de Fabry.
La thérapie génique, qui consiste à introduire le gène GLA via un vecteur viral adéno-associé (AAV) pour produire de manière endogène l’α-Gal A, est considérée comme un traitement curatif prometteur.
La thérapie de réduction du substrat (SRT) est une approche qui inhibe la synthèse du Gb3 elle-même. Contrairement à l’ERT, elle est développée comme un médicament oral ne nécessitant pas d’administration intraveineuse.
L’expansion du dépistage néonatal est également un enjeu important. Une intervention thérapeutique précoce grâce à un diagnostic précoce pourrait améliorer le pronostic à long terme3).
Les connaissances sur la diversité phénotypique s’accumulent également. Il a été rapporté que même avec la même mutation GLA, des tableaux cliniques allant de la forme classique à la forme limitée à un organe peuvent se présenter1), et que le profil d’inactivation du chromosome X chez les femmes conductrices ne prédit pas le phénotype. Dans les rapports de cas de maladie de Fabry non classique se manifestant principalement par des symptômes neurologiques, certains cas ont été diagnostiqués après un examen approfondi des lésions de la substance blanche cérébrale2).
Furia A, Ditaranto R, Biagini E, et al. Fabry disease in W162C mutation: a case report of two patients and a review of literature. BMC Neurol. 2024;24:113. PMCID: PMC10996216. doi:10.1186/s12883-024-03540-3.
Ferreira Tátá C, Massas M, Pinto F, Caçador N, Silva AL. Fabry Disease: A Atypical Presentation. Cureus. 2021;13(10):e18708. doi:10.7759/cureus.18708. PMID:34790463; PMCID:PMC8582620.
Starcea IM, Bodescu Amancei Ionescu L, Lazaruc TI, Lupu VV, Bogos RA, Ioniuc I, et al. Palm-Plant Pain, Sign of a Severe Systemic Disease? Case Report and Review of Literature. Genes. 2023;14(2). doi:10.3390/genes14020516. PMID:36833443; PMCID:PMC9957027.
Yanfang W, Juanjuan H, Shengli Z, Lei Y, Fei G. Case report and literature review: Fabry disease misdiagnosing as polymyalgia rheumatica. Medicine. 2023;102(44):e34630. doi:10.1097/MD.0000000000034630. PMID:37933054; PMCID:PMC10627660.
Copiez le texte de l'article et collez-le dans l'assistant IA de votre choix.
Article copié dans le presse-papiers
Ouvrez un assistant IA ci-dessous et collez le texte copié dans la conversation.