A doença de Fabry é uma doença metabólica hereditária causada pela deficiência de α-galactosidase A (α-Gal A), uma enzima hidrolase lisossômica, resultando no acúmulo de globotriaosilceramida (Gb3) e outros glicoesfingolipídios nas células de todo o corpo.
Apresenta herança recessiva ligada ao X devido a mutações no gene GLA (Xq22.1). Foi relatada independentemente por Anderson e Fabry em 18982). A prevalência é estimada em cerca de 1 em 10.000 pessoas, mas a triagem neonatal relata frequências mais altas (1:3.000 a 1:7.800)3).
O quadro clínico é dividido em tipo clássico e tipo de início tardio (não clássico). No tipo clássico, a atividade da α-Gal A está quase ausente, e o dano multiorgânico aparece desde a infância. No tipo de início tardio, há atividade enzimática residual, e a doença pode se manifestar na idade adulta como dano a um único órgão, como coração ou rim1, 2).
Tipo
Época de início
Principais órgãos afetados
Clássico
Infância até idade escolar
Múltiplos órgãos (sistêmico)
Tipo tardio
Idade adulta
Coração e rins
QA doença de Fabry é hereditária?
A
Sim, é uma doença genética ligada ao cromossomo X com herança recessiva. Todas as filhas de um paciente do sexo masculino serão portadoras, e não é transmitida aos filhos. As portadoras do sexo feminino podem apresentar uma ampla gama de sintomas, desde assintomáticas até graves, dependendo do padrão de inativação do cromossomo X. Se houver alguém na sua família com esta doença, recomenda-se aconselhamento genético.
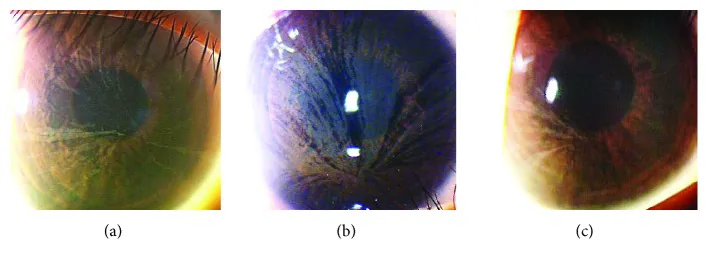
Yasuhito Ikegawa, Atsushi Shiraishi, Yasuhito Hayashi, Akiyoshi Ogimoto, et al. In Vivo Confocal Microscopic Observations of Vortex Keratopathy in Patients with Amiodarone-Induced Keratopathy and Fabry Disease 2018 Mar 21 J Ophthalmol. 2018 Mar 21; 2018:5315137 Figure 4. PMCID: PMC5884153. License: CC BY.
Achados do exame com lâmpada de fenda de três pacientes com doença de Fabry. (a) Achados da mãe de 32 anos. (b) Achados da filha de 8 anos. (c) Achados da filha de 4 anos.
A diminuição da acuidade visual devido à opacidade corneana em espiral é rara. Na maioria dos casos, as queixas subjetivas relacionadas aos sintomas oculares são escassas.
Como sintomas sistêmicos, a dor em queimação nas extremidades (acroparestesia) é o sintoma mais precoce. A diminuição da sudorese (hipoidrose) e os sintomas gastrointestinais também aparecem desde a infância.
Idade de início: Observada a partir de cerca de 6 anos
Localização: No nível da membrana basal do epitélio corneano
Achado: Depósitos acinzentados pálidos que se espalham em espiral por toda a córnea
Portadores: Também observada com frequência em portadoras heterozigóticas do sexo feminino
Catarata de Fabry
Localização: subcapsular posterior e córtex do cristalino
Achado: opacidade em forma de raios de roda (spoke-like) característica
Impacto na visão: geralmente leve, mas em casos avançados pode indicar cirurgia de catarata
Alterações vasculares
Vasos conjuntivais: apresentam-se tortuosos e dilatados
Vasos retinianos: Caracteriza-se por tortuosidade das veias principais em ambos os olhos
Microaneurismas: Podem ser observados na conjuntiva e na retina
Na forma tardia ou cardíaca, os achados típicos da córnea e da pele podem estar ausentes. Em um caso de doença de Fabry cardíaca com mutação W162C, foi relatado que a avaliação oftalmológica não revelou opacidade corneal nem angioceratoma1).
QO padrão espiralado da córnea afeta a visão?
A
A opacidade corneal (cornea verticillata) é um depósito na camada superficial do epitélio da córnea e geralmente não afeta a visão. Frequentemente é descoberta pela primeira vez no exame com lâmpada de fenda, e raramente é percebida pelo próprio paciente. No entanto, se a catarata progredir, pode ocorrer diminuição da visão.
A causa da doença de Fabry é uma mutação no gene GLA. Mais de 1000 tipos de mutações foram relatados, e o quadro clínico varia muito dependendo do tipo de mutação 2).
Por ser uma herança recessiva ligada ao X, os homens (hemizigotos) tendem a ser mais graves. As mulheres portadoras (heterozigotas) também apresentam uma ampla variedade de quadros clínicos, desde assintomáticas até graves como a forma clássica, devido ao desvio da inativação do cromossomo X 1). Cerca de 1% das mulheres portadoras apresentam sintomas, e a confirmação da opacidade corneana é útil para triagem tanto em homens quanto em mulheres.
O principal fator de risco é o histórico familiar da doença de Fabry. Em pacientes com dor inexplicável nas extremidades, acidente vascular cerebral em idade jovem, cardiomegalia inexplicável ou insuficiência renal inexplicável, esta doença deve ser considerada no diagnóstico diferencial 2, 4).
O exame com lâmpada de fenda é o mais importante. A córnea verticilada é um achado característico e serve como pista para suspeitar desta doença. A presença de catarata (opacidade axial subcapsular posterior) também deve ser verificada. Em casos não clássicos, os achados oftalmológicos podem estar ausentes1), portanto, a ausência de achados não deve excluir a doença.
Em homens, o diagnóstico definitivo é possível pela medição da atividade da enzima α-Gal A. Em mulheres portadoras, a atividade enzimática pode estar dentro da faixa normal devido à inativação do X, sendo necessária a análise genética 2). A Lyso-Gb3 plasmática é um biomarcador com alta sensibilidade diagnóstica, também útil na triagem de mulheres 4).
O diagnóstico diferencial mais importante para a córnea verticilada é a deposição corneana medicamentosa causada pela amiodarona (fármaco antiarrítmico). A opacidade decorrente do uso de amiodarona apresenta achados muito semelhantes à córnea verticilada da doença de Fabry. A verificação do histórico de medicação é a chave para o diagnóstico diferencial.
Além disso, é necessário diferenciar de outras deposições corneanas medicamentosas, como cloroquina e indometacina, e de outras doenças que causam córnea verticilada.
O tratamento definitivo para a doença de Fabry é a terapia de reposição enzimática (enzyme replacement therapy: ERT). Agalsidase alfa (Replagal®) ou agalsidase beta (Fabrazyme®) são administrados por infusão intravenosa a cada duas semanas. A ERT pode reduzir a dor nas extremidades, retardar o declínio da função renal e inibir a progressão da hipertrofia cardíaca. O início precoce é importante para prevenir a progressão do dano aos órgãos.
Migalastate (Galafold®) é um medicamento oral indicado para pacientes com mutações passíveis de tratamento. Ele estabiliza a estrutura tridimensional da proteína α-Gal A desnaturada, facilitando seu transporte para o lisossomo.
Não é necessário tratamento específico para a opacidade corneana em vórtice. Se a catarata progredir e afetar a função visual, a cirurgia de catarata está indicada.
QO tratamento melhora os sintomas da córnea?
A
A terapia de reposição enzimática (TRE) é eficaz em retardar a progressão dos sintomas renais, cardíacos e neurológicos, mas geralmente não melhora os depósitos corneanos em espiral. No entanto, como esses depósitos não afetam significativamente a acuidade visual, não é necessário tratamento agressivo. Se a catarata afetar a visão, a cirurgia é realizada.
Na doença de Fabry, a mutação no gene GLA causa redução ou perda da atividade da α-Gal A. Como resultado, o Gb3 e sua forma desacilada, Lyso-Gb3, que deveriam ser degradados por essa enzima, acumulam-se nos lisossomos. O Gb3 acumulado causa disfunção celular, ativação de cascatas inflamatórias e estresse oxidativo, levando a danos progressivos em múltiplos órgãos.
Na córnea, o Gb3 se acumula nas células-tronco basais do limbo. À medida que essas células migram centrifugamente, formam um padrão de depósito em espiral (córnea verticillata). O depósito ocorre no nível da membrana basal do epitélio corneano.
No cristalino, o Gb3 se acumula nas células epiteliais e fibras do córtex, causando opacidade em forma de raios (spoke-like) sob a cápsula posterior. Esta é a chamada catarata de Fabry.
O acúmulo de Gb3 nas células endoteliais vasculares e células musculares lisas causa tortuosidade e dilatação dos vasos conjuntivais e retinianos. Na retina, é observado como tortuosidade venosa principal bilateral. Sistemicamente, o acúmulo nos podócitos glomerulares renais e cardiomiócitos causa dano renal e hipertrofia cardíaca.
Além da terapia de reposição enzimática (ERT) convencional, várias novas abordagens terapêuticas estão sendo estudadas para a doença de Fabry.
A terapia gênica é uma terapia curativa esperada para produzir α-Gal A endogenamente através da introdução do gene GLA usando vetor de vírus adeno-associado (AAV) ou outros.
Terapia de Inibição da Síntese de Substrato (SRT) é uma abordagem que inibe a síntese de Gb3 em si, e ao contrário da ERT, está sendo desenvolvida como um medicamento oral que não requer administração intravenosa.
A expansão da triagem neonatal também é uma questão importante. A intervenção precoce através do diagnóstico precoce pode melhorar o prognóstico a longo prazo3).
O conhecimento sobre a diversidade fenotípica também está se acumulando. Foi relatado que a mesma mutação GLA pode apresentar diferentes quadros clínicos, desde o tipo clássico até o tipo limitado a órgãos1), e que o padrão de inativação do cromossomo X em mulheres portadoras não prediz o fenótipo. Em relatos de casos com sintomas neurológicos da doença de Fabry não clássica como queixa principal, há casos em que o diagnóstico da doença de Fabry foi feito através de exame detalhado de lesões da substância branca cerebral2).
Furia A, Ditaranto R, Biagini E, et al. Fabry disease in W162C mutation: a case report of two patients and a review of literature. BMC Neurol. 2024;24:113. PMCID: PMC10996216. doi:10.1186/s12883-024-03540-3.
Ferreira Tátá C, Massas M, Pinto F, Caçador N, Silva AL. Fabry Disease: A Atypical Presentation. Cureus. 2021;13(10):e18708. doi:10.7759/cureus.18708. PMID:34790463; PMCID:PMC8582620.
Starcea IM, Bodescu Amancei Ionescu L, Lazaruc TI, Lupu VV, Bogos RA, Ioniuc I, et al. Palm-Plant Pain, Sign of a Severe Systemic Disease? Case Report and Review of Literature. Genes. 2023;14(2). doi:10.3390/genes14020516. PMID:36833443; PMCID:PMC9957027.
Yanfang W, Juanjuan H, Shengli Z, Lei Y, Fei G. Case report and literature review: Fabry disease misdiagnosing as polymyalgia rheumatica. Medicine. 2023;102(44):e34630. doi:10.1097/MD.0000000000034630. PMID:37933054; PMCID:PMC10627660.
Copie o texto do artigo e cole no assistente de IA de sua preferência.
Artigo copiado para a área de transferência
Abra um assistente de IA abaixo e cole o texto copiado na conversa.