Болезнь Фабри — это наследственное метаболическое заболевание, вызванное дефицитом лизосомного гидролитического фермента α-галактозидазы A (α-Gal A), что приводит к накоплению сфингогликолипидов, включая глоботриаозилцерамид (Gb3), в клетках всего организма.
Заболевание наследуется по X-сцепленному рецессивному типу, обусловлено мутациями в гене GLA (Xq22.1). Независимо описано Андерсоном и Фабри в 1898 году 2). Распространенность оценивается примерно как 1 на 10 000 человек, однако при неонатальном скрининге сообщается о более высокой частоте (1:3 000–1:7 800) 3).
Клиническая картина делится на классический тип и поздний (неклассический) тип. При классическом типе активность α-Gal A практически отсутствует, и с детства появляются множественные поражения органов. При позднем типе сохраняется остаточная ферментативная активность, и заболевание может проявиться во взрослом возрасте как изолированное поражение сердца или почек 1, 2).
Тип заболевания
Возраст начала
Основные пораженные органы
Классический
Младенчество – школьный возраст
Мультиорганный (системный)
Поздняя форма
Взрослый возраст
Сердце и почки
QНаследуется ли болезнь Фабри?
A
Да, это X-сцепленное рецессивное заболевание. Все дочери мужчины-пациента являются носителями, а сыновья не наследуют заболевание. Женщины-носители могут иметь широкий спектр симптомов — от бессимптомных до тяжелых — в зависимости от паттерна инактивации X-хромосомы. Если у вас в семье есть больные этим заболеванием, рекомендуется пройти генетическое консультирование.
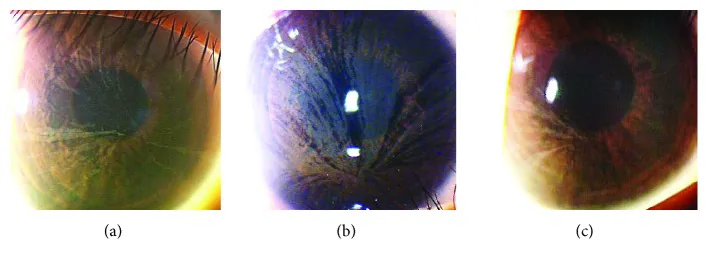
Yasuhito Ikegawa, Atsushi Shiraishi, Yasuhito Hayashi, Akiyoshi Ogimoto, et al. In Vivo Confocal Microscopic Observations of Vortex Keratopathy in Patients with Amiodarone-Induced Keratopathy and Fabry Disease 2018 Mar 21 J Ophthalmol. 2018 Mar 21; 2018:5315137 Figure 4. PMCID: PMC5884153. License: CC BY.
Результаты биомикроскопии с щелевой лампой трех пациентов с болезнью Фабри. (a) Результаты 32-летней матери. (b) Результаты 8-летней дочери. (c) Результаты 4-летней дочери.
Снижение остроты зрения из-за спиралевидного помутнения роговицы встречается редко. Во многих случаях субъективные жалобы на глазные симптомы отсутствуют.
К системным симптомам относятся жгучие боли в конечностях (акропарестезия) как самый ранний симптом. Снижение потоотделения (гипогидроз) и желудочно-кишечные симптомы также появляются с детства.
Локализация: на уровне базальной мембраны эпителия роговицы
Признаки: бледно-серые пигментные отложения, распространяющиеся по всей роговице в виде завихрений
Носители: также часто наблюдается у женщин-гетерозиготных носителей
Катаракта Фабри
Локализация : задняя капсула и кора хрусталика
Признак : характерные спицеобразные помутнения
Влияние на зрение : обычно незначительное, но в запущенных случаях может потребоваться хирургия катаракты
Сосудистые изменения
Конъюнктивальные сосуды : извитость и расширение
Сосуды сетчатки: характерна двусторонняя извитость основных вен
Микроаневризмы: могут наблюдаться в конъюнктиве и сетчатке
При поздних или кардиальных формах типичные признаки роговицы и кожи могут отсутствовать. В случае кардиальной болезни Фабри с мутацией W162C при офтальмологическом обследовании не было выявлено ни вертициллятной кератопатии, ни ангиокератом1).
QВлияют ли завитки роговицы на зрение?
A
Вертициллятная кератопатия (cornea verticillata) представляет собой отложения в поверхностных слоях эпителия роговицы и обычно не влияет на зрение. Часто впервые обнаруживается при осмотре с помощью щелевой лампы, и сам пациент почти никогда ее не замечает. Однако при прогрессировании катаракты может возникнуть снижение остроты зрения.
Причиной болезни Фабри является мутация гена GLA. Сообщается о более чем 1000 различных мутациях, и клиническая картина значительно варьирует в зависимости от типа мутации2).
Поскольку это X-сцепленное рецессивное наследование, мужчины (гемизиготы) более склонны к тяжелому течению. Женщины-носители (гетерозиготы) также демонстрируют разнообразную клиническую картину — от бессимптомного течения до тяжелой классической формы — из-за неравномерной инактивации X-хромосомы1). Примерно у 1% женщин-носителей развиваются симптомы, и выявление помутнения роговицы полезно для скрининга как у мужчин, так и у женщин.
Основным фактором риска является семейный анамнез болезни Фабри. У пациентов с необъяснимыми акральными болями, ювенильным инсультом, необъяснимой гипертрофией сердца или необъяснимым поражением почек это заболевание должно быть включено в дифференциальный диагноз2, 4).
Биомикроскопия с щелевой лампой является наиболее важной. Вортициформная роговица является характерным признаком и служит поводом заподозрить это заболевание. Также проверьте наличие катаракты (задняя субкапсулярная спицеобразная помутнение). В неклассических случаях офтальмологические признаки могут отсутствовать 1); отсутствие признаков не должно исключать это заболевание.
У мужчин окончательный диагноз может быть установлен путем измерения активности α-Gal A. У женщин-носителей из-за инактивации X-хромосомы активность фермента может находиться в пределах нормы, поэтому требуется генетический анализ 2). Плазменный Lyso-Gb3 является биомаркером с высокой диагностической чувствительностью, также полезным для скрининга у женщин 4).
Наиболее важным дифференциальным диагнозом завитой роговицы является лекарственное отложение в роговице, вызванное амиодароном (антиаритмическим препаратом). Помутнения, возникающие при приеме амиодарона, очень похожи на завитую роговицу при болезни Фабри. Подтверждение анамнеза приема лекарств является ключом к дифференциальной диагностике.
Кроме того, необходима дифференциация от других лекарственных отложений в роговице, таких как хлорохин, индометацин, а также от других заболеваний, вызывающих cornea verticillata.
Основным методом лечения болезни Фабри является ферментозаместительная терапия (enzyme replacement therapy: ERT). Агалсидаза альфа (Replagal®) или агалсидаза бета (Fabrazyme®) вводятся внутривенно капельно каждые две недели. ФЗТ позволяет уменьшить боль в конечностях, замедлить снижение функции почек и подавить прогрессирование гипертрофии сердца. Раннее начало важно для замедления прогрессирования поражения органов.
Мигаластат (Галафолд®) — пероральный препарат, показанный пациентам с аменабельными мутациями. Он стабилизирует трехмерную структуру денатурированного белка α-Gal A и способствует его транспорту в лизосому.
Специфического лечения для завиткового помутнения роговицы не требуется. При прогрессировании катаракты, влияющей на зрительную функцию, показана операция по удалению катаракты.
QУлучшает ли лечение симптомы роговицы?
A
Ферментозаместительная терапия (ФЗТ) замедляет прогрессирование почечных, сердечных и неврологических симптомов, но обычно не улучшает завитковые отложения в роговице. Однако, поскольку эти роговичные изменения существенно не влияют на зрение, агрессивное лечение не требуется. Если катаракта влияет на зрение, проводится хирургическое вмешательство.
При болезни Фабри мутации гена GLA приводят к снижению или потере активности α-Gal A. В результате Gb3 и его деацилированная форма Lyso-Gb3, которые в норме должны расщепляться этим ферментом, накапливаются в лизосомах. Накопление Gb3 вызывает клеточную дисфункцию, активацию воспалительных каскадов и окислительный стресс, что приводит к прогрессирующему поражению многих органов.
В роговице Gb3 накапливается в базальных стволовых клетках лимба. При центробежной миграции этих клеток формируется завитковый рисунок отложений (cornea verticillata). Отложения происходят на уровне базальной мембраны эпителия роговицы.
В хрусталике Gb3 накапливается в эпителиальных клетках и кортикальных волокнах, вызывая помутнение в виде спиц (spoke-like) под задней капсулой. Это называется катарактой Фабри.
Накопление Gb3 в эндотелиальных клетках сосудов и гладкомышечных клетках вызывает извитость и расширение сосудов конъюнктивы и сетчатки. В сетчатке это наблюдается как двусторонняя извитость основных вен. Системно накопление в подоцитах почечных клубочков и кардиомиоцитах приводит к повреждению почек и гипертрофии сердца.
Помимо традиционной ERT, изучается несколько новых терапевтических подходов к болезни Фабри.
Генная терапия, при которой ген GLA вводится с помощью аденоассоциированного вирусного (AAV) вектора для эндогенной продукции α-Gal A, рассматривается как перспективное радикальное лечение.
Терапия ингибирования синтеза субстрата (SRT) — это подход, подавляющий синтез Gb3. В отличие от ERT, она разрабатывается как пероральный препарат, не требующий внутривенного введения.
Расширение неонатального скрининга также является важной задачей. Раннее терапевтическое вмешательство благодаря ранней диагностике может улучшить долгосрочный прогноз3).
Накоплены также знания о фенотипическом разнообразии. Сообщается, что даже при одной и той же мутации GLA могут наблюдаться различные клинические картины от классической до органоограниченной формы1), и что паттерн инактивации Х-хромосомы у женщин-носителей не предсказывает фенотип. В описаниях случаев неклассической болезни Фабри с преимущественно неврологическими симптомами были примеры, когда диагноз болезни Фабри был поставлен после тщательного исследования поражений белого вещества головного мозга2).
Furia A, Ditaranto R, Biagini E, et al. Fabry disease in W162C mutation: a case report of two patients and a review of literature. BMC Neurol. 2024;24:113. PMCID: PMC10996216. doi:10.1186/s12883-024-03540-3.
Ferreira Tátá C, Massas M, Pinto F, Caçador N, Silva AL. Fabry Disease: A Atypical Presentation. Cureus. 2021;13(10):e18708. doi:10.7759/cureus.18708. PMID:34790463; PMCID:PMC8582620.
Starcea IM, Bodescu Amancei Ionescu L, Lazaruc TI, Lupu VV, Bogos RA, Ioniuc I, et al. Palm-Plant Pain, Sign of a Severe Systemic Disease? Case Report and Review of Literature. Genes. 2023;14(2). doi:10.3390/genes14020516. PMID:36833443; PMCID:PMC9957027.
Yanfang W, Juanjuan H, Shengli Z, Lei Y, Fei G. Case report and literature review: Fabry disease misdiagnosing as polymyalgia rheumatica. Medicine. 2023;102(44):e34630. doi:10.1097/MD.0000000000034630. PMID:37933054; PMCID:PMC10627660.
Скопируйте текст статьи и вставьте его в выбранный ИИ-ассистент.
Статья скопирована в буфер обмена
Откройте ИИ-ассистент ниже и вставьте скопированный текст в чат.