โรคฟาบรีเป็น โรคสะสมในไลโซโซมที่ถ่ายทอดทางโครโมโซม X เกิดจาก การขาดเอนไซม์ α-กาแลกโตซิเดส A (α-Gal A)
กระจกตา ลายเกลียว (cornea verticillata)อาการทางตานอกจากกระจกตา ลายเกลียว ได้แก่ ต้อกระจก ฟาบรี (ฝ้าขุ่นใต้แคปซูลด้านหลังเป็นรูปซี่ล้อ)การคดเคี้ยวของหลอดเลือดเยื่อบุตา และจอประสาทตา
การบำบัดด้วยเอนไซม์ทดแทน (ERT) สามารถชะลอการดำเนินของอาการทั่วร่างกายได้ แต่การสะสมที่กระจกตา มักจะยังคงอยู่พบความผิดปกติที่กระจกตา ในหญิงที่เป็นพาหะเฮเทอโรไซกัสด้วย ซึ่งเป็นเบาะแสสำคัญในการคัดกรอง
โรคฟาบรีเป็นโรคเมตาบอลิกทางพันธุกรรมที่เกิดจากการขาดเอนไซม์อัลฟา-กาแลกโตซิเดส เอ (α-Gal A) ซึ่งเป็นเอนไซม์ไฮโดรเลสในไลโซโซม ส่งผลให้เกิดการสะสมของโกลโบไทรโอซิลเซราไมด์ (Gb3) และไกลโคสฟิงโกลิปิดอื่นๆ ในเซลล์ทั่วร่างกาย
โรคนี้มีการถ่ายทอดทางพันธุกรรมแบบด้อยสัมพันธ์กับโครโมโซม X เนื่องจากการกลายพันธุ์ของยีน GLA (Xq22.1) รายงานครั้งแรกโดย Anderson และ Fabry อย่างอิสระในปี ค.ศ. 18982) ความชุกประมาณ 1 ใน 10,000 คน แต่การตรวจคัดกรองทารกแรกเกิดรายงานความถี่ที่สูงกว่า (1:3,000 ถึง 1:7,800)3)
ภาพทางคลินิกแบ่งออกเป็น ชนิดคลาสสิก และ ชนิดเริ่มมีอาการช้า (ไม่ใช่คลาสสิก) ในชนิดคลาสสิก กิจกรรมของ α-Gal A เกือบจะหายไป และเกิดความเสียหายต่อหลายอวัยวะตั้งแต่วัยเด็ก ในชนิดเริ่มมีอาการช้า ยังมีกิจกรรมของเอนไซม์เหลืออยู่ และโรคอาจแสดงออกในวัยผู้ใหญ่เป็นความเสียหายต่ออวัยวะเดียว เช่น หัวใจหรือไต1, 2)
ชนิด ช่วงเวลาที่เริ่มมีอาการ อวัยวะหลักที่ได้รับผลกระทบ ชนิดคลาสสิก วัยเด็กตอนต้นถึงวัยเรียน หลายอวัยวะ (ทั่วร่างกาย) ชนิดที่เริ่มมีอาการช้า วัยผู้ใหญ่ หัวใจและไต
Q
โรคฟาบรีเป็นโรคทางพันธุกรรมหรือไม่?
A
ใช่ เป็นโรคทางพันธุกรรมแบบถอยที่เชื่อมโยงกับโครโมโซม X บุตรสาวทั้งหมดของผู้ป่วยชายจะเป็นพาหะของโรค และไม่ถ่ายทอดไปยังบุตรชาย พาหะหญิงอาจมีอาการตั้งแต่ไม่มีอาการไปจนถึงอาการรุนแรง ขึ้นอยู่กับรูปแบบการหยุดการทำงานของโครโมโซม X หากมีสมาชิกในครอบครัวเป็นโรคนี้ แนะนำให้รับคำปรึกษาทางพันธุกรรม
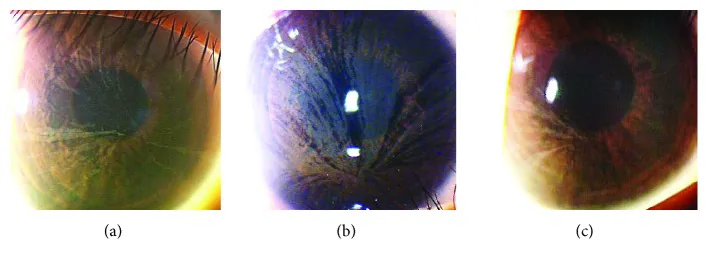
Yasuhito Ikegawa, Atsushi Shiraishi, Yasuhito Hayashi, Akiyoshi Ogimoto, et al. In Vivo Confocal Microscopic Observations of Vortex Keratopathy in Patients with Amiodarone-Induced Keratopathy and Fabry Disease 2018 Mar 21 J Ophthalmol. 2018 Mar 21; 2018:5315137 Figure 4. PMCID: PMC5884153. License: CC BY.
ผลการตรวจด้วยกล้องจุลทรรศน์ชีวภาพแบบร่องกราดของผู้ป่วยโรคฟาบรีสามราย (ก) ผลการตรวจของมารดาอายุ 32 ปี (ข) ผลการตรวจของบุตรสาวอายุ 8 ปี (ค) ผลการตรวจของบุตรสาวอายุ 4 ปี
การมองเห็น ลดลงจากความขุ่นของกระจกตา แบบเกลียวนั้นพบได้น้อย ในหลายกรณี อาการทางตาที่ผู้ป่วยรับรู้ได้มีน้อย
อาการทั่วร่างกาย อาการปวดแสบร้อนที่ปลายแขนปลายขา (acroparesthesia) เป็นอาการที่พบเร็วที่สุด เหงื่อออกน้อย (ภาวะเหงื่อน้อย) และอาการทางระบบทางเดินอาหารก็ปรากฏตั้งแต่วัยเด็ก
กระจกตาเป็นเกลียว (cornea verticillata)
ความถี่ : พบในผู้ป่วย 50–80%
อายุที่เริ่มพบ : สังเกตได้ตั้งแต่อายุประมาณ 6 ปี
ตำแหน่ง : ที่ระดับเยื่อฐานของเยื่อบุกระจกตา
ลักษณะที่พบ : การสะสมของเม็ดสีเทาจางๆ กระจายเป็นเกลียวทั่วกระจกตา
พาหะ : พบบ่อยในเพศหญิงที่เป็นพาหะเฮเทอโรไซกัสด้วย
ต้อกระจกฟาบรี
ตำแหน่ง : ใต้แคปซูลด้านหลังและเนื้อเลนส์
ลักษณะ : ความขุ่นเป็นรูปซี่ล้อ (spoke-like) ที่เป็นลักษณะเฉพาะ
ผลต่อการมองเห็น : โดยทั่วไปเล็กน้อย แต่ในกรณีที่ลุกลามอาจเป็นข้อบ่งชี้ในการผ่าตัดต้อกระจก
การเปลี่ยนแปลงของหลอดเลือด
หลอดเลือดเยื่อบุตา : พบว่าคดเคี้ยวและขยายตัว
หลอดเลือดจอตา : มีลักษณะเฉพาะคือเส้นเลือดดำหลักคดเคี้ยวในตาทั้งสองข้าง
หลอดเลือดโป่งพองขนาดเล็ก เยื่อบุตา และจอตา
ในชนิดที่เกิดช้าหรือชนิดจำกัดเฉพาะหัวใจ อาจไม่มีลักษณะทั่วไปที่กระจกตา และผิวหนัง ในกรณีโรคฟาบรีชนิดหัวใจที่มีการกลายพันธุ์ W162C มีรายงานว่าการประเมินทางจักษุวิทยาไม่พบความขุ่นของกระจกตา หรือแองจิโอเคอราโตมา1) .
Q
ลายเกลียวที่กระจกตาส่งผลต่อการมองเห็นหรือไม่?
A
ความขุ่นของกระจกตา (cornea verticillata) คือการสะสมในชั้นผิวของเยื่อบุกระจกตา และโดยปกติแล้วไม่ส่งผลต่อการมองเห็น มักพบครั้งแรกจากการตรวจด้วยกล้องจุลทรรศน์ชนิดกรีด และผู้ป่วยแทบจะไม่สังเกตเห็นด้วยตนเอง อย่างไรก็ตาม หากต้อกระจก ลุกลาม อาจทำให้การมองเห็น ลดลง
สาเหตุของโรคฟาบรีคือการกลายพันธุ์ของยีน GLA มีรายงานการกลายพันธุ์มากกว่า 1,000 ชนิด และลักษณะทางคลินิกแตกต่างกันอย่างมากตามชนิดของการกลายพันธุ์ 2) .
เนื่องจากเป็นโรคถ่ายทอดทางพันธุกรรมแบบด้อยที่เชื่อมโยงกับโครโมโซม X เพศชาย (hemizygous) จึงมีแนวโน้มที่จะมีอาการรุนแรงกว่า เพศหญิงที่เป็นพาหะ (heterozygous) ก็มีลักษณะทางคลินิกที่หลากหลาย ตั้งแต่ไม่มีอาการไปจนถึงรุนแรงแบบคลาสสิก เนื่องจากการเบี่ยงเบนของการหยุดทำงานของโครโมโซม X 1) พบว่าประมาณ 1% ของเพศหญิงที่เป็นพาหะมีอาการ และการยืนยันความขุ่นของกระจกตา มีประโยชน์ในการคัดกรองทั้งในเพศชายและเพศหญิง
ปัจจัยเสี่ยงหลักคือประวัติครอบครัวเป็นโรคฟาบรี ในผู้ป่วยที่มีอาการปวดปลายแขนขาโดยไม่ทราบสาเหตุ โรคหลอดเลือดสมองในวัยหนุ่มสาว ภาวะหัวใจโตโดยไม่ทราบสาเหตุ หรือภาวะไตเสื่อมโดยไม่ทราบสาเหตุ ควรพิจารณาโรคนี้ในการวินิจฉัยแยกโรค 2, 4) .
โรคฟาบรีไม่สามารถป้องกันได้เนื่องจากเป็นโรคทางพันธุกรรม แต่หากมีประวัติครอบครัว แนะนำให้ตรวจทางพันธุกรรมตั้งแต่เนิ่นๆ การตรวจพบตั้งแต่เนิ่นๆ และการเริ่มการรักษาตั้งแต่เนิ่นๆ มีความสำคัญในการชะลอการดำเนินของความเสียหายต่ออวัยวะ การหลีกเลี่ยงสภาพแวดล้อมที่มีอุณหภูมิสูงและการรักษาความชุ่มชื้นที่เพียงพอช่วยจัดการกับภาวะเหงื่อออกน้อย
การตรวจด้วยกล้องจุลทรรศน์ชนิดกรีด (Slit-lamp)กระจกตา แบบวน (vortex cornea) เป็นลักษณะเฉพาะที่ทำให้สงสัยโรคนี้ ควรตรวจสอบว่ามีต้อกระจก (ความขุ่นตามแนวแกนใต้แคปซูลด้านหลัง) หรือไม่ ในกรณีที่ไม่ใช่แบบคลาสสิก อาจไม่มีผลการตรวจทางตา1) ดังนั้นการไม่มีผลการตรวจไม่ควรตัดโรคนี้ทิ้ง
รายการตรวจ ความสำคัญในการวินิจฉัย กิจกรรมของ α-Gal A ยืนยันในเพศชาย Lyso-Gb3 ตัวบ่งชี้ทางชีวภาพ การวิเคราะห์ยีน GLA การระบุการกลายพันธุ์
ในเพศชาย การวินิจฉัยที่แน่นอนสามารถทำได้โดยการวัดกิจกรรมของเอนไซม์ α-Gal A ในเพศหญิงที่เป็นพาหะ กิจกรรมของเอนไซม์อาจอยู่ในช่วงปกติเนื่องจากการหยุดการทำงานของโครโมโซม X จึงจำเป็นต้องมีการวิเคราะห์ทางพันธุกรรม 2) Lyso-Gb3 ในพลาสมาเป็นตัวบ่งชี้ทางชีวภาพ ที่มีความไวในการวินิจฉัยสูง และยังมีประโยชน์ในการคัดกรองเพศหญิง 4)
การวินิจฉัยแยกโรคที่สำคัญที่สุดสำหรับกระจกตา แบบเกลียวคือ การสะสมของยาในกระจกตา ที่เกิดจากอะมิโอดาโรน (ยาต้านจังหวะการเต้นของหัวใจ) ความขุ่นที่เกิดจากการรับประทานอะมิโอดาโรนมีลักษณะคล้ายคลึงกับกระจกตา แบบเกลียวในโรคฟาบรีมาก การตรวจสอบประวัติการใช้ยาเป็นกุญแจสำคัญในการวินิจฉัยแยกโรค
นอกจากนี้ จำเป็นต้องแยกโรคจากการสะสมของยาในกระจกตา จากคลอโรควิน อินโดเมธาซิน และโรคอื่นๆ ที่ทำให้เกิด กระจกตาแบบเกลียว
การรักษาที่เป็นสาเหตุของโรคฟาบรีคือ การบำบัดด้วยเอนไซม์ทดแทน (enzyme replacement therapy: ERT) ให้อะกัลซิเดส อัลฟา (Replagal®) หรืออะกัลซิเดส เบตา (Fabrazyme®) โดยการหยดทางหลอดเลือดดำทุกสองสัปดาห์ ERT สามารถลดอาการปวดปลายแขนขา ชะลอการเสื่อมของไต และยับยั้งการลุกลามของภาวะหัวใจโต การเริ่มต้นเร็วมีความสำคัญในการป้องกันความเสียหายของอวัยวะที่ลุกลาม
มิกาลาสแตท (Galafold®) เป็นยารับประทานที่ใช้ในผู้ป่วยที่มีการกลายพันธุ์แบบ amenable โดยจะทำให้โครงสร้างสามมิติของโปรตีน α-Gal A ที่เสียสภาพคงตัว และส่งเสริมการขนส่งไปยังไลโซโซม
ไม่จำเป็นต้องรักษาเฉพาะสำหรับความขุ่นของกระจกตา แบบ vortex หากต้อกระจก ดำเนินไปและส่งผลต่อการมองเห็น การผ่าตัดต้อกระจก เป็นข้อบ่งชี้
Q
การรักษาจะช่วยให้อาการทางกระจกตาดีขึ้นหรือไม่?
A
การบำบัดด้วยเอนไซม์ทดแทน (ERT) มีประสิทธิภาพในการชะลอการดำเนินของอาการทางไต หัวใจ และระบบประสาท แต่โดยทั่วไปแล้วไม่สามารถทำให้การสะสมแบบเกลียวในกระจกตา ดีขึ้น อย่างไรก็ตาม เนื่องจากการสะสมในกระจกตา ไม่ส่งผลต่อการมองเห็น อย่างมีนัยสำคัญ จึงไม่จำเป็นต้องรักษาเชิงรุก หากต้อกระจก ส่งผลต่อการมองเห็น จะทำการผ่าตัด
ในโรคฟาบรี การกลายพันธุ์ของยีน GLA ทำให้การทำงานของ α-Gal A ลดลงหรือหายไป ส่งผลให้ Gb3 และรูปแบบที่ถูกกำจัดหมู่ acyl คือ Lyso-Gb3 ซึ่งเอนไซม์นี้ควรจะย่อยสลาย สะสมในไลโซโซม Gb3 ที่สะสมทำให้เกิดความผิดปกติของเซลล์ การกระตุ้น cascade การอักเสบ และภาวะเครียดออกซิเดชัน นำไปสู่ความเสียหายหลายอวัยวะแบบค่อยเป็นค่อยไป
ในกระจกตา Gb3 สะสมในเซลล์ต้นกำเนิดฐานของลิมบัส เมื่อเซลล์เหล่านี้เคลื่อนที่ออกจากศูนย์กลาง จะเกิดรูปแบบการสะสมแบบเกลียว (cornea verticillata) การสะสมเกิดขึ้นที่ระดับเยื่อฐานของเยื่อบุกระจกตา
ในเลนส์ตา Gb3 จะสะสมในเซลล์เยื่อบุผิวและเส้นใยคอร์เทกซ์ ทำให้เกิดความขุ่นคล้ายซี่ล้อ (spoke-like) ใต้แคปซูลด้านหลัง ซึ่งเรียกว่าต้อกระจก ฟาบรี
การสะสมของ Gb3 ในเซลล์บุผนังหลอดเลือดและเซลล์กล้ามเนื้อเรียบทำให้เกิดการคดเคี้ยวและขยายของหลอดเลือดเยื่อบุตา และจอประสาทตา ในจอประสาทตา จะสังเกตเห็นเป็นการคดเคี้ยวของหลอดเลือดดำหลักทั้งสองข้าง ในระดับระบบ การสะสมในพอดอไซต์ของโกลเมอรูลัสไตและเซลล์กล้ามเนื้อหัวใจทำให้เกิดความเสียหายของไตและหัวใจโต
นอกเหนือจากการบำบัดทดแทนเอนไซม์ (ERT) แบบดั้งเดิมแล้ว ยังมีการศึกษาวิธีการรักษาแบบใหม่หลายวิธีสำหรับโรคฟาบรี
การบำบัดด้วยยีน
การบำบัดด้วยการยับยั้งการสังเคราะห์ซับสเตรต (SRT) เป็นแนวทางที่ยับยั้งการสังเคราะห์ Gb3 เอง และแตกต่างจาก ERT ตรงที่กำลังถูกพัฒนาเป็นยารับประทานที่ไม่ต้องให้ทางหลอดเลือดดำ
การขยายการตรวจคัดกรองทารกแรกเกิด ก็เป็นประเด็นสำคัญเช่นกัน การแทรกแซงการรักษาตั้งแต่เนิ่นๆ ผ่านการวินิจฉัยตั้งแต่แรกเริ่มอาจช่วยปรับปรุงการพยากรณ์โรคในระยะยาว3)
ความรู้เกี่ยวกับความหลากหลายของฟีโนไทป์ก็กำลังสะสมเช่นกัน มีรายงานว่าการกลายพันธุ์ GLA เดียวกันสามารถแสดงภาพทางคลินิกที่แตกต่างกันตั้งแต่แบบคลาสสิกไปจนถึงแบบจำกัดเฉพาะอวัยวะ1) และรูปแบบการหยุดการทำงานของโครโมโซม X ในเพศหญิงที่เป็นพาหะไม่สามารถทำนายฟีโนไทป์ได้ ในรายงานผู้ป่วยที่มีอาการทางระบบประสาทของโรคฟาบรีที่ไม่ใช่แบบคลาสสิกเป็นอาการหลัก มีกรณีที่ได้รับการวินิจฉัยว่าเป็นโรคฟาบรีผ่านการตรวจสอบรอยโรคของเนื้อขาวในสมองอย่างละเอียด2)
บทความนี้มีวัตถุประสงค์เพื่อให้ข้อมูลสำหรับบุคลากรทางการแพทย์ และไม่ใช่สิ่งทดแทนการวินิจฉัยหรือการรักษา โปรดปรึกษาแพทย์ผู้เชี่ยวชาญด้านจักษุวิทยาและเมแทบอลิซึมเพื่อการวินิจฉัยและรักษาโรคฟาบรี ช่วงเวลาในการเริ่มการบำบัดด้วยเอนไซม์ทดแทนและการเลือกวิธีการรักษาจะพิจารณาตามสภาพของผู้ป่วยแต่ละราย
Furia A, Ditaranto R, Biagini E, et al. Fabry disease in W162C mutation: a case report of two patients and a review of literature. BMC Neurol. 2024;24:113. PMCID: PMC10996216. doi:10.1186/s12883-024-03540-3.
Ferreira Tátá C, Massas M, Pinto F, Caçador N, Silva AL. Fabry Disease: A Atypical Presentation. Cureus. 2021;13(10):e18708. doi:10.7759/cureus.18708. PMID:34790463; PMCID:PMC8582620.
Starcea IM, Bodescu Amancei Ionescu L, Lazaruc TI, Lupu VV, Bogos RA, Ioniuc I, et al. Palm-Plant Pain, Sign of a Severe Systemic Disease? Case Report and Review of Literature. Genes. 2023;14(2). doi:10.3390/genes14020516. PMID:36833443; PMCID:PMC9957027.
Yanfang W, Juanjuan H, Shengli Z, Lei Y, Fei G. Case report and literature review: Fabry disease misdiagnosing as polymyalgia rheumatica. Medicine. 2023;102(44):e34630. doi:10.1097/MD.0000000000034630. PMID:37933054; PMCID:PMC10627660.