La enfermedad de Fabry es un trastorno metabólico hereditario causado por una deficiencia de la hidrolasa lisosomal α-galactosidasa A (α-Gal A), que conduce a la acumulación de glucoesfingolípidos, incluida la globotriaosilceramida (Gb3), en las células de todo el cuerpo.
Presenta herencia recesiva ligada al cromosoma X debido a mutaciones en el gen GLA (Xq22.1). Fue reportada de forma independiente por Anderson y Fabry en 1898 2). La prevalencia se estima en aproximadamente 1 de cada 10,000, pero los cribados neonatales reportan frecuencias más altas (1:3,000 a 1:7,800) 3).
Las manifestaciones clínicas se dividen ampliamente en tipo clásico y tardío (no clásico). En el tipo clásico, la actividad de α-Gal A está casi ausente y el daño multiorgánico aparece desde la primera infancia. En el tipo tardío, hay actividad enzimática residual y puede presentarse como afectación de un solo órgano (corazón o riñón) en la edad adulta 1, 2).
Tipo de enfermedad
Edad de inicio
Órganos principales afectados
Tipo clásico
Infancia a edad escolar
Múltiples órganos (sistémico)
Tipo de inicio tardío
Edad adulta
Corazón y riñones
Q¿La enfermedad de Fabry es hereditaria?
A
Sí, es un trastorno hereditario recesivo ligado al cromosoma X. Todas las hijas de un paciente varón son portadoras, mientras que los hijos no heredan la enfermedad. Las portadoras femeninas pueden presentar una amplia gama de síntomas, desde asintomáticas hasta graves, según el patrón de inactivación del cromosoma X. Si tiene un familiar con esta enfermedad, se recomienda asesoramiento genético.
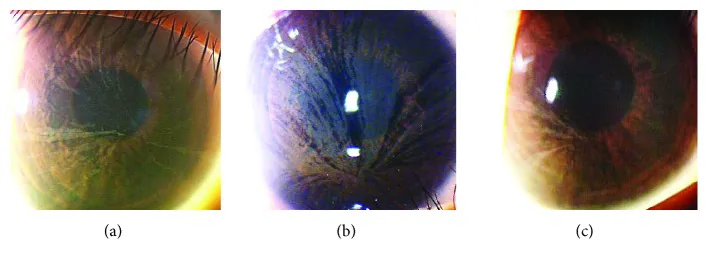
Yasuhito Ikegawa, Atsushi Shiraishi, Yasuhito Hayashi, Akiyoshi Ogimoto, et al. In Vivo Confocal Microscopic Observations of Vortex Keratopathy in Patients with Amiodarone-Induced Keratopathy and Fabry Disease 2018 Mar 21 J Ophthalmol. 2018 Mar 21; 2018:5315137 Figure 4. PMCID: PMC5884153. License: CC BY.
Hallazgos de biomicroscopía con lámpara de hendidura en tres pacientes con enfermedad de Fabry. (a) Hallazgos en una madre de 32 años. (b) Hallazgos en una hija de 8 años. (c) Hallazgos en una hija de 4 años.
La disminución de la agudeza visual debido a la opacidad corneal en remolino es rara. En muchos casos, las quejas subjetivas relacionadas con los síntomas oculares son escasas.
Los síntomas sistémicos incluyen dolor urente en las extremidades (acroparestesia) como el síntoma más temprano. La disminución de la sudoración (hipohidrosis) y los síntomas gastrointestinales también aparecen en la infancia.
Frecuencia: Presente en el 50–80% de los pacientes
Edad de inicio: Observable desde aproximadamente los 6 años
Ubicación: A nivel de la membrana basal del epitelio corneal
Hallazgos: Pigmentación grisácea tenue que se extiende en forma de remolino por toda la córnea
Portadores: También se observa con frecuencia en portadoras heterocigotas femeninas
Catarata de Fabry
Ubicación: Subcapsular posterior y corteza del cristalino
Hallazgos: Opacidades características en forma de radios de rueda
Efecto sobre la visión: Generalmente leve, pero los casos avanzados pueden requerir cirugía de cataratas
Cambios vasculares
Vasos conjuntivales: Se observan tortuosidad y dilatación
Vasos retinianos: Tortuosidad bilateral característica de las venas retinianas principales
Microaneurismas: Pueden observarse en la conjuntiva y la retina
En las formas de inicio tardío o limitadas al corazón, pueden faltar los hallazgos típicos corneales y cutáneos. En un caso de enfermedad de Fabry cardíaca con la mutación W162C, la evaluación oftalmológica no reveló ni córnea verticilada ni angiokeratomas 1).
Q¿El patrón en espiral de la córnea afecta la visión?
A
La córnea verticilada (cornea verticillata) es un depósito en la capa superficial del epitelio corneal y generalmente no afecta la visión. A menudo se descubre por primera vez mediante microscopía con lámpara de hendidura y rara vez es notada por el paciente. Sin embargo, si las cataratas progresan, puede ocurrir pérdida de visión.
La enfermedad de Fabry es causada por mutaciones en el gen GLA. Se han reportado más de 1000 tipos de mutaciones, y la presentación clínica varía mucho según el tipo de mutación 2).
Debido a que es un trastorno recesivo ligado al cromosoma X, los varones (hemicigotos) tienden a presentar síntomas graves. Las mujeres portadoras (heterocigotas) también presentan una amplia gama de manifestaciones clínicas, desde asintomáticas hasta formas clásicas graves, debido a la inactivación sesgada del cromosoma X 1). Se estima que aproximadamente el 1% de las portadoras desarrollan síntomas, y la opacidad corneal es útil para el cribado en ambos sexos.
El principal factor de riesgo son los antecedentes familiares de enfermedad de Fabry. Los pacientes con acroparestesias inexplicables, accidente cerebrovascular juvenil, hipertrofia cardíaca inexplicable o enfermedad renal inexplicable deben considerar esta enfermedad en el diagnóstico diferencial 2, 4).
La microscopía con lámpara de hendidura es la más importante. La queratopatía en vórtice es un hallazgo característico y proporciona una pista para sospechar esta enfermedad. También se debe confirmar la presencia de cataratas (opacidad subcapsular posterior en forma de radios). En casos no clásicos, los hallazgos oftalmológicos pueden estar ausentes 1); por lo tanto, la ausencia de hallazgos no descarta esta enfermedad.
En los hombres, el diagnóstico definitivo se puede realizar midiendo la actividad de la enzima α-Gal A. En las mujeres portadoras, la actividad enzimática puede estar dentro del rango normal debido a la inactivación del cromosoma X, por lo que es necesario un análisis genético 2). La Lyso-Gb3 plasmática es un biomarcador con alta sensibilidad diagnóstica y también es útil para el cribado en mujeres 4).
El diagnóstico diferencial más importante para la queratopatía en vórtice es la deposición corneal inducida por amiodarona (un fármaco antiarrítmico). Las opacidades resultantes del uso de amiodarona se asemejan mucho a la queratopatía en vórtice de la enfermedad de Fabry. Confirmar el historial de medicación es clave para la diferenciación.
Además, es necesaria la diferenciación de los depósitos corneales inducidos por fármacos como cloroquina, indometacina y otros, así como de otras enfermedades que causan córnea verticilada.
El tratamiento fundamental para la enfermedad de Fabry es la terapia de reemplazo enzimático (TRE). Se administra agalsidasa alfa (Replagal®) o agalsidasa beta (Fabrazyme®) por vía intravenosa cada dos semanas. Se espera que la TRE reduzca el dolor acroparestésico, retrase el deterioro de la función renal y suprima la progresión de la hipertrofia cardíaca. El inicio temprano es importante para suprimir la progresión del daño orgánico.
Migalastat (Galafold®) es un medicamento oral indicado para pacientes con mutaciones susceptibles. Estabiliza la conformación de la proteína α-Gal A mutada y facilita su transporte a los lisosomas.
No se requiere tratamiento específico para la queratopatía en vórtice. Si la catarata progresa y afecta la función visual, puede estar indicada la cirugía de cataratas.
Q¿El tratamiento mejora los síntomas corneales?
A
La terapia de reemplazo enzimático (TRE) puede retrasar la progresión de los síntomas renales, cardíacos y neurológicos, pero generalmente no mejora los depósitos corneales en forma de remolino. Sin embargo, dado que los hallazgos corneales no afectan significativamente la visión, no es necesario un tratamiento activo. Se realiza cirugía si las cataratas afectan la visión.
En la enfermedad de Fabry, las mutaciones en el gen GLA reducen o eliminan la actividad de la α-Gal A. Como resultado, el Gb3 y su forma desacilada, Lyso-Gb3, que esta enzima normalmente descompone, se acumulan en los lisosomas. El Gb3 acumulado causa disfunción celular, activación de cascadas inflamatorias y estrés oxidativo, lo que lleva a daño multiorgánico progresivo.
En la córnea, el Gb3 se acumula en las células madre basales del limbo. A medida que estas células migran centrífugamente, forman un patrón de depósito en forma de remolino (córnea verticillata). Los depósitos ocurren a nivel de la membrana basal del epitelio corneal.
En el cristalino, Gb3 se acumula en las células epiteliales y fibras corticales, causando opacidades en forma de radios bajo la cápsula posterior. Este hallazgo se denomina catarata de Fabry.
La acumulación de Gb3 en las células endoteliales vasculares y células musculares lisas causa tortuosidad y dilatación de los vasos conjuntivales y retinianos. En la retina, se observa como tortuosidad venosa principal bilateral. Sistémicamente, la acumulación en podocitos de los glomérulos renales y cardiomiocitos conduce a daño renal e hipertrofia cardíaca.
7. Investigación más reciente y perspectivas futuras
Además de la ERT convencional, se están estudiando varios enfoques terapéuticos novedosos para la enfermedad de Fabry.
La terapia génica se espera como un tratamiento curativo que introduce el gen GLA mediante vectores de virus adenoasociados (AAV) para producir α-Gal A endógena.
La terapia de reducción de sustrato (SRT) es un enfoque que suprime la síntesis de Gb3 en sí misma y, a diferencia de la ERT, se está desarrollando como un fármaco oral que no requiere administración intravenosa.
La expansión del cribado neonatal también es un tema importante. El diagnóstico temprano y la intervención temprana pueden mejorar el pronóstico a largo plazo3).
También se está acumulando conocimiento sobre la diversidad fenotípica. Se ha informado que incluso con la misma mutación GLA, pueden presentarse diferentes cuadros clínicos que van desde la forma clásica hasta la limitada a órganos1), y que los patrones de inactivación del cromosoma X en mujeres portadoras no predicen el fenotipo. En informes de casos de enfermedad de Fabry no clásica que se presentan con síntomas neurológicos, hay ejemplos en los que el examen detallado de las lesiones de la sustancia blanca condujo al diagnóstico de la enfermedad de Fabry2).
Furia A, Ditaranto R, Biagini E, et al. Fabry disease in W162C mutation: a case report of two patients and a review of literature. BMC Neurol. 2024;24:113. PMCID: PMC10996216. doi:10.1186/s12883-024-03540-3.
Ferreira Tátá C, Massas M, Pinto F, Caçador N, Silva AL. Fabry Disease: A Atypical Presentation. Cureus. 2021;13(10):e18708. doi:10.7759/cureus.18708. PMID:34790463; PMCID:PMC8582620.
Starcea IM, Bodescu Amancei Ionescu L, Lazaruc TI, Lupu VV, Bogos RA, Ioniuc I, et al. Palm-Plant Pain, Sign of a Severe Systemic Disease? Case Report and Review of Literature. Genes. 2023;14(2). doi:10.3390/genes14020516. PMID:36833443; PMCID:PMC9957027.
Yanfang W, Juanjuan H, Shengli Z, Lei Y, Fei G. Case report and literature review: Fabry disease misdiagnosing as polymyalgia rheumatica. Medicine. 2023;102(44):e34630. doi:10.1097/MD.0000000000034630. PMID:37933054; PMCID:PMC10627660.
Copia el texto del artículo y pégalo en el asistente de IA que prefieras.
Artículo copiado al portapapeles
Abre un asistente de IA abajo y pega el texto copiado en el chat.