مرض فابري هو اضطراب استقلابي وراثي ناتج عن نقص إنزيم ألفا-جلاكتوزيداز A (α-Gal A)، وهو إنزيم محلل للجسيمات الحالة، مما يؤدي إلى تراكم الغلوبوترياوسيلسيراميد (Gb3) وغيره من السفينغوليبيدات السكرية في خلايا الجسم المختلفة.
يظهر المرض نمطًا وراثيًا متنحيًا مرتبطًا بالكروموسوم X بسبب طفرات في جين GLA (Xq22.1). تم الإبلاغ عنه بشكل مستقل من قبل أندرسون وفابري في عام 18982). يُقدر معدل الانتشار بحوالي 1 من كل 10,000 شخص، لكن فحص حديثي الولادة أشار إلى تواتر أعلى (1:3,000 إلى 1:7,800)3).
تنقسم الصورة السريرية إلى النمط الكلاسيكي والنمط المتأخر (غير الكلاسيكي). في النمط الكلاسيكي، يكون نشاط α-Gal A منعدمًا تقريبًا، وتظهر إصابة متعددة الأعضاء منذ الطفولة المبكرة. في النمط المتأخر، يوجد نشاط إنزيمي متبقٍ، وقد يظهر المرض في مرحلة البلوغ كإصابة عضو واحد مثل القلب أو الكلى1, 2).
النوع
وقت الظهور
الأعضاء الرئيسية المتضررة
النمط الكلاسيكي
الطفولة المبكرة إلى سن المدرسة
أعضاء متعددة (الجهاز بأكمله)
النوع المتأخر
مرحلة البلوغ
القلب والكلى
Qهل مرض فابري وراثي؟
A
نعم، هو مرض وراثي متنحي مرتبط بالكروموسوم X. جميع بنات المريض الذكر يصبحن حاملات للمرض، ولا يورث للأبناء. قد تظهر على الحاملات الأنثى أعراض تتراوح من عدم ظهور أعراض إلى أعراض شديدة، وذلك حسب نمط تعطيل الكروموسوم X. إذا كان أحد أفراد عائلتك مصابًا بهذا المرض، ننصحك بالحصول على استشارة وراثية.
Yasuhito Ikegawa, Atsushi Shiraishi, Yasuhito Hayashi, Akiyoshi Ogimoto, et al. In Vivo Confocal Microscopic Observations of Vortex Keratopathy in Patients with Amiodarone-Induced Keratopathy and Fabry Disease 2018 Mar 21 J Ophthalmol. 2018 Mar 21; 2018:5315137 Figure 4. PMCID: PMC5884153. License: CC BY.
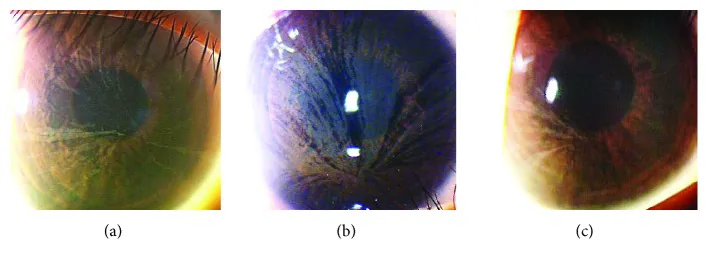
نتائج فحص المصباح الشقي لثلاثة مرضى بمرض فابري. (أ) نتائج أم عمرها 32 عامًا. (ب) نتائج ابنة عمرها 8 أعوام. (ج) نتائج ابنة عمرها 4 أعوام.
المنظر: ترسبات رمادية شاحبة تنتشر بشكل حلزوني عبر القرنية بأكملها
الناقلون: يُلاحظ أيضًا بشكل متكرر لدى الناقلات المتغايرات الإناث
إعتام عدسة فابري
الموقع: تحت المحفظة الخلفية وقشرة العدسة
العلامات: عتامة تشبه قضبان العجلة (spoke-like) مميزة
تأثير على الرؤية: عادةً طفيف، لكن في الحالات المتقدمة قد يستدعي جراحة الساد
تغيرات الأوعية الدموية
أوعية الملتحمة: تظهر متعرجة ومتوسعة
الأوعية الدموية الشبكية: يتميز بتعرج الأوردة الرئيسية في كلتا العينين
تمدد الأوعية الدموية الدقيقة: قد يظهر في الملتحمة والشبكية
في النوع المتأخر أو المقتصر على القلب، قد تغيب العلامات النمطية في القرنية والجلد. في حالة مرض فابري القلبي مع طفرة W162C، تم الإبلاغ عن عدم وجود تورد القرنية أو الورم الوعائي القرني في التقييم العيني1).
Qهل يؤثر النمط الحلزوني للقرنية على الرؤية؟
A
تورد القرنية (cornea verticillata) هو ترسب في الطبقة السطحية من ظهارة القرنية، وعادة لا يؤثر على الرؤية. غالبًا ما يُكتشف لأول مرة بفحص المصباح الشقي، ونادرًا ما يلاحظه المريض بنفسه. ومع ذلك، إذا تقدم إعتام عدسة العين، فقد يحدث انخفاض في الرؤية.
سبب مرض فابري هو طفرة في جين GLA. تم الإبلاغ عن أكثر من 1000 نوع من الطفرات، وتختلف الصورة السريرية بشكل كبير حسب نوع الطفرة 2).
نظرًا لأنه وراثة متنحية مرتبطة بـ X، فإن الذكور (متماثلي الزيجوت) أكثر عرضة للإصابة الشديدة. كما تظهر الإناث الحاملات (متغايرات الزيجوت) مجموعة متنوعة من الصور السريرية، بدءًا من عدم ظهور الأعراض إلى الحالات الشديدة المشابهة للنمط الكلاسيكي، بسبب انحراف تعطيل الكروموسوم X 1). يُقدر أن حوالي 1% من الحاملات تظهر عليهن الأعراض، ويعتبر تأكيد عتامة القرنية مفيدًا في الفحص لكل من الذكور والإناث.
عامل الخطر الرئيسي هو التاريخ العائلي لمرض فابري. يجب النظر في هذا المرض لدى المرضى الذين يعانون من آلام غير مفسرة في الأطراف، أو سكتة دماغية في سن مبكرة، أو تضخم القلب غير المفسر، أو اعتلال الكلى غير المفسر 2, 4).
فحص المصباح الشقي هو الأكثر أهمية. القرنية الحلزونية هي علامة مميزة وتكون سببًا للاشتباه في هذا المرض. يجب أيضًا التحقق من وجود إعتام عدسة العين (عتامة محورية تحت المحفظة الخلفية). في الحالات غير التقليدية، قد تغيب النتائج العينية1)، لذا لا ينبغي استبعاد المرض بسبب غياب النتائج.
عند الذكور، يمكن تأكيد التشخيص عن طريق قياس نشاط إنزيم α-Gal A. أما عند الإناث الحاملات، فقد يكون نشاط الإنزيم ضمن النطاق الطبيعي بسبب تعطيل X، مما يستدعي التحليل الجيني 2). يُعد Lyso-Gb3 في البلازما مؤشرًا حيويًا عالي الحساسية للتشخيص، ومفيدًا أيضًا في فحص الإناث 4).
أهم تشخيص تفريقي لاعتلال القرنية الحلزوني هو ترسبات القرنية الدوائية الناتجة عن الأميودارون (دواء مضاد لاضطراب النظم). العتامة الناتجة عن تناول الأميودارون تشبه إلى حد كبير اعتلال القرنية الحلزوني في مرض فابري. يُعد التحقق من تاريخ تناول الدواء مفتاحًا للتشخيص التفريقي.
بالإضافة إلى ذلك، يجب التفريق بين ترسبات القرنية الدوائية الناتجة عن الكلوروكين والإندوميتاسين وغيرها، وبين الأمراض الأخرى التي تسبب قرنية حلزونية.
العلاج الأساسي لمرض فابري هو العلاج بالإنزيم البديل (ERT). يتم إعطاء أجالسيداز ألفا (ريبليجال®) أو أجالسيداز بيتا (فابرازيم®) عن طريق التسريب الوريدي كل أسبوعين. يُتوقع من ERT تخفيف آلام الأطراف، وإبطاء تدهور وظائف الكلى، وتثبيط تقدم تضخم القلب. البدء المبكر مهم لمنع تقدم تلف الأعضاء.
ميجالاستات (جالافولد®) هو دواء فموي، ويُستخدم للمرضى الذين لديهم طفرات قابلة للعلاج. يعمل على تثبيت البنية الثلاثية لبروتين α-Gal A المتغير، مما يعزز نقله إلى الليزوزوم.
لا حاجة لعلاج محدد لعتامة القرنية الحلزونية. إذا تقدم إعتام عدسة العين وأثر على الوظيفة البصرية، يُوصى بإجراء جراحة الساد.
Qهل تتحسن أعراض القرنية بالعلاج؟
A
العلاج بالإنزيم البديل (ERT) فعال في إبطاء تقدم الأعراض الكلوية والقلبية والعصبية، لكنه لا يحسن عادةً الترسبات القرنية الحلزونية. ومع ذلك، نظرًا لأن هذه الترسبات لا تؤثر بشكل كبير على حدة البصر، فلا حاجة لعلاج فعال. إذا كان إعتام عدسة العين يؤثر على الرؤية، يتم إجراء الجراحة.
في مرض فابري، يؤدي طفرة جين GLA إلى انخفاض أو فقدان نشاط إنزيم α-Gal A. ونتيجة لذلك، يتراكم كل من Gb3 ومشتقه منزوع الأسيل Lyso-Gb3 داخل الليزوزومات. يسبب Gb3 المتراكم خللًا وظيفيًا خلويًا، وتنشيط سلسلة الالتهاب، والإجهاد التأكسدي، مما يؤدي إلى تلف تدريجي متعدد الأعضاء.
في القرنية، يتراكم Gb3 في الخلايا الجذعية القاعدية للحوف. أثناء هجرة هذه الخلايا نابذة، تشكل نمط الترسبات الحلزونية (cornea verticillata). يحدث الترسيب على مستوى الغشاء القاعدي للظهارة القرنية.
في العدسة، يتراكم Gb3 في الخلايا الظهارية وألياف القشرة، مما يؤدي إلى عتامة تشبه المتحدث (spoke-like) تحت المحفظة الخلفية. هذه هي ما يُسمى بإعتام عدسة فابري.
يؤدي تراكم Gb3 في الخلايا البطانية للأوعية الدموية وخلايا العضلات الملساء إلى تمدد وتعرج الأوعية الدموية في الملتحمة والشبكية. في الشبكية، يُلاحظ على شكل تعرج ثنائي للأوردة الرئيسية. بشكل جهازي، يؤدي التراكم في الخلايا الرجلية (podocytes) للكبيبات الكلوية وخلايا عضلة القلب إلى تلف الكلى وتضخم القلب.
بالإضافة إلى العلاج التقليدي بالإنزيم التعويضي (ERT)، يتم دراسة عدة طرق علاجية جديدة لمرض فابري.
العلاج الجيني هو علاج جذري يُتوقع أن ينتج α-Gal A داخليًا عن طريق إدخال جين GLA باستخدام ناقل فيروسي مرتبط بالأدينو (AAV) أو غيره.
علاج تثبيط تخليق الركيزة (SRT) هو نهج يثبط تخليق Gb3 نفسه، وعلى عكس ERT، يتم تطويره كدواء فموي لا يتطلب إعطاء وريدي.
توسيع فحص حديثي الولادة هو أيضًا قضية مهمة. قد يؤدي التدخل العلاجي المبكر من خلال التشخيص المبكر إلى تحسين التشخيص طويل الأمد3).
كما تتراكم المعرفة حول تنوع النمط الظاهري. تم الإبلاغ عن أن نفس طفرة GLA يمكن أن تظهر صورًا سريرية مختلفة تتراوح من الشكل الكلاسيكي إلى الشكل المحدود بالأعضاء1)، وأن نمط تعطيل الكروموسوم X لدى الناقلات الإناث لا يتنبأ بالنمط الظاهري. في تقارير الحالات التي يكون فيها العرض الرئيسي هو الأعراض العصبية لمرض فابري غير الكلاسيكي، هناك حالات تم فيها تشخيص مرض فابري من خلال الفحص الدقيق لآفات المادة البيضاء في الدماغ2).
Furia A, Ditaranto R, Biagini E, et al. Fabry disease in W162C mutation: a case report of two patients and a review of literature. BMC Neurol. 2024;24:113. PMCID: PMC10996216. doi:10.1186/s12883-024-03540-3.
Ferreira Tátá C, Massas M, Pinto F, Caçador N, Silva AL. Fabry Disease: A Atypical Presentation. Cureus. 2021;13(10):e18708. doi:10.7759/cureus.18708. PMID:34790463; PMCID:PMC8582620.
Starcea IM, Bodescu Amancei Ionescu L, Lazaruc TI, Lupu VV, Bogos RA, Ioniuc I, et al. Palm-Plant Pain, Sign of a Severe Systemic Disease? Case Report and Review of Literature. Genes. 2023;14(2). doi:10.3390/genes14020516. PMID:36833443; PMCID:PMC9957027.
Yanfang W, Juanjuan H, Shengli Z, Lei Y, Fei G. Case report and literature review: Fabry disease misdiagnosing as polymyalgia rheumatica. Medicine. 2023;102(44):e34630. doi:10.1097/MD.0000000000034630. PMID:37933054; PMCID:PMC10627660.
انسخ نص المقال والصقه في مساعد الذكاء الاصطناعي الذي تفضله.
تم نسخ المقال إلى الحافظة
افتح أحد مساعدي الذكاء الاصطناعي أدناه والصق النص المنسوخ في مربع المحادثة.