涡状角膜(cornea verticillata)
频率:出现在50%~80%的患者中
发病年龄:从6岁左右开始出现
部位:角膜上皮基底膜水平
表现:灰白色淡色素沉着呈漩涡状扩散至整个角膜
携带者:女性杂合子携带者也常见
法布里病是一种遗传性代谢疾病,由于溶酶体水解酶α-半乳糖苷酶A(α-Gal A)缺乏,导致包括三己糖酰基鞘脂醇(Gb3)在内的鞘糖脂在全身细胞中蓄积。
由GLA基因(Xq22.1)突变引起X连锁隐性遗传。1898年由Anderson和Fabry分别报道2)。患病率估计约为1/10,000,但新生儿筛查报告更高频率(1:3,000至1:7,800)3)。
临床表现大致分为经典型和迟发型(非经典型)。经典型中α-Gal A活性几乎消失,从幼儿期开始出现多器官损害。迟发型中残留酶活性,可能在成年期表现为心脏或肾脏的单器官受累1, 2)。
| 病型 | 发病时期 | 主要受累器官 |
|---|---|---|
| 经典型 | 婴幼儿至学龄期 | 多器官(全身) |
| 迟发型 | 成年期 | 心脏和肾脏 |
是的,这是一种X连锁隐性遗传病。男性患者的女儿均为携带者,而儿子不会遗传。女性携带者因X染色体失活模式不同,可能从无症状到重症表现各异。如果您的家人患有此病,建议进行遗传咨询。
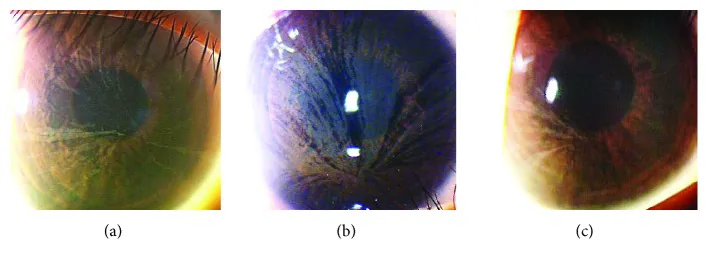
角膜涡状混浊导致的视力下降很少见。多数情况下,眼部症状的主观主诉很少。
全身症状包括四肢末端的灼痛(肢端感觉异常)是最早的症状。出汗减少(少汗症)和胃肠道症状也在儿童期出现。
涡状角膜(cornea verticillata)
频率:出现在50%~80%的患者中
发病年龄:从6岁左右开始出现
部位:角膜上皮基底膜水平
表现:灰白色淡色素沉着呈漩涡状扩散至整个角膜
携带者:女性杂合子携带者也常见
法布里白内障
血管系统变化
结膜血管:可见迂曲和扩张
视网膜血管:特征性双眼主干静脉迂曲
微动脉瘤:可见于结膜和视网膜
迟发型或心脏局限型可能缺乏典型的角膜和皮肤表现。据报道,一例携带W162C突变的心脏型法布里病患者,眼科评估未发现角膜涡状混浊或血管角化瘤1)。
法布里病是由GLA基因突变引起的。已报道超过1000种突变,突变类型不同,临床表现差异很大2)。
由于是X连锁隐性遗传病,男性(半合子)症状往往较重。女性携带者(杂合子)由于X染色体失活偏斜,也可表现出从无症状到接近经典型的严重症状等多种临床表现1)。约1%的女性携带者会出现症状,角膜混浊对男女筛查均有用。
主要风险因素是法布里病的家族史。对于原因不明的肢端疼痛、青年卒中、原因不明的心肌肥厚或原因不明的肾损害患者,需将本病纳入鉴别诊断2, 4)。
裂隙灯显微镜检查最为重要。涡状角膜是特征性表现,是怀疑本病的契机。还需确认是否存在白内障(后囊下辐轮状混浊)。非经典型病例可能缺乏眼科表现1),因此不能因无表现而排除本病。
| 检查项目 | 诊断意义 |
|---|---|
| α-Gal A活性 | 男性确诊 |
| Lyso-Gb3 | 生物标志物 |
| GLA基因分析 | 突变鉴定 |
男性可通过测量α-Gal A酶活性进行确诊。女性携带者由于X失活的影响,酶活性可能显示正常范围,因此需要进行基因分析2)。血浆Lyso-Gb3是一种诊断灵敏度高的生物标志物,对女性筛查也有用4)。
涡状角膜最重要的鉴别诊断是**胺碘酮(抗心律失常药)**引起的药物性角膜沉积症。服用胺碘酮产生的混浊与法布里病的涡状角膜极为相似。确认用药史是鉴别的关键。
此外,还需要与氯喹、吲哚美辛等药物引起的角膜沉积,以及其他导致角膜涡状混浊的疾病进行鉴别。
法布里病的根本治疗是酶替代疗法(ERT)。每两周静脉滴注阿加糖酶α(Replagal®)或阿加糖酶β(Fabrazyme®)。ERT有望减轻肢端疼痛、延缓肾功能下降、抑制心脏肥大的进展。早期开始治疗对于抑制器官损伤的进展至关重要。
米加司他(Galafold®)是一种口服药物,适用于具有可治疗突变的患者。它能稳定变性的α-Gal A蛋白的立体结构,促进其向溶酶体的转运。
涡状角膜病变无需特殊治疗。如果白内障进展并影响视功能,则可考虑白内障手术。
酶替代疗法(ERT)可抑制肾脏、心脏和神经症状的进展,但通常不能改善角膜的漩涡状沉积。然而,由于角膜表现对视力影响不大,因此无需积极治疗。如果白内障影响视力,则进行手术。
法布里病中,GLA基因突变导致α-半乳糖苷酶A活性降低或丧失。结果,该酶本应分解的Gb3及其脱酰基形式Lyso-Gb3在溶酶体中积累。积累的Gb3引起细胞功能障碍、炎症级联反应激活和氧化应激,导致进行性多器官损伤。
在角膜中,Gb3积聚在角膜缘的基底干细胞中。这些细胞在离心迁移过程中形成漩涡状沉积模式(角膜涡状混浊)。沉积发生在角膜上皮基底膜水平。
在晶状体中,Gb3积聚在上皮细胞和皮质纤维中,导致后囊下出现辐轮状混浊。这一发现被称为法布里白内障。
血管内皮细胞和平滑肌细胞中的Gb3积聚导致结膜和视网膜血管迂曲扩张。在视网膜中,表现为双眼主干静脉迂曲。全身性方面,肾小球足细胞和心肌细胞中的积聚导致肾损伤和心脏肥大。
除了传统的ERT,法布里病的多种新型治疗方法正在研究中。
基因治疗通过腺相关病毒(AAV)载体导入GLA基因,产生内源性α-Gal A,有望成为一种根治性治疗方法。
**底物合成抑制疗法(SRT)**是一种抑制Gb3本身合成的方法,与ERT不同,它正在开发为一种无需静脉给药的口服药物。
扩大新生儿筛查也是一个重要课题。早期诊断和早期治疗干预可能改善长期预后3)。
关于表型多样性的知识也在积累。据报道,即使具有相同的GLA突变,也可能出现从经典型到器官局限型的不同临床表现1),并且女性携带者中的X染色体失活模式不能预测表型。在以神经系统症状为主诉的非经典型法布里病病例报告中,有通过详细检查脑白质病变而诊断出法布里病的例子2)。