La sindrome di Anton è una condizione di anosognosia visiva in cui i pazienti con cecità corticale non riconoscono la propria perdita della vista e riferiscono esperienze visive inesistenti. I pazienti, nonostante siano ciechi, tentano di descrivere dettagliatamente ciò che vedono, producendo confabulazioni.
La malattia prende il nome dal neurologo austriaco Gabriel Anton. Anton riportò un caso di una donna di 69 anni con lesioni temporali bilaterali che presentava anosognosia acquisita e sordità. Il termine “anosognosia” fu coniato da Joseph Babinski, per cui la sindrome è anche chiamata “sindrome di Anton-Babinski”. La prima descrizione di anosognosia visiva risale allo schiavo romano Arpaste, che negava la propria cecità e sosteneva che la stanza fosse buia.
Differenza dalla sindrome di Charles Bonnet: è importante notare che nella sindrome di Charles Bonnet i pazienti con deficit visivo sperimentano allucinazioni visive, ma mantengono la consapevolezza del proprio deficit visivo, differenziandosi fondamentalmente dalla sindrome di Anton.
Epidemiologia: l’età mediana è di 55 anni (range 6-96 anni), senza differenze di genere. Il caso più frequente è secondario a un accidente cerebrovascolare (CVA), comune negli anziani con multipli fattori di rischio vascolare. L’ictus dell’arteria cerebrale posteriore (PCA) rappresenta il 5-10% di tutti gli ictus. 1) Tra il 1965 e il 2016 sono stati riportati solo 28 casi di sindrome di Anton-Babinski, rendendola una malattia estremamente rara. 2)
QQual è la differenza tra la sindrome di Anton e la sindrome di Charles Bonnet?
A
La sindrome di Anton è caratterizzata da negazione della cecità e confabulazione in pazienti con cecità corticale, con mancanza di insight. Al contrario, nella sindrome di Charles Bonnet i pazienti con deficit visivo sperimentano allucinazioni visive, ma mantengono l’insight del proprio deficit visivo. Entrambe hanno alla base un disturbo visivo, ma differiscono fondamentalmente per la presenza o assenza di anosognosia.
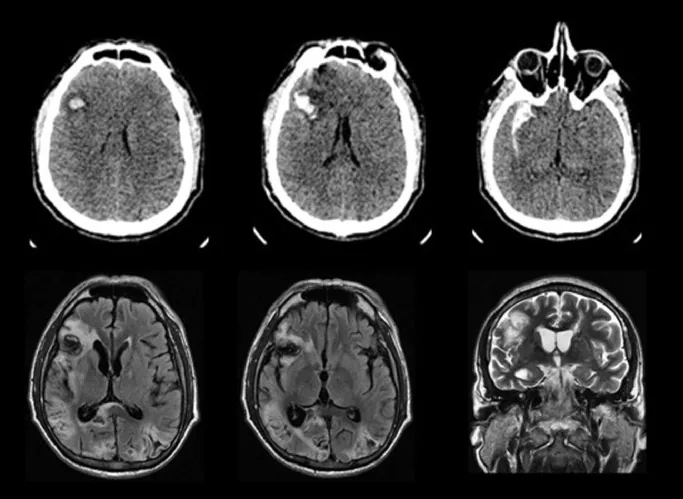
Immagini TC e RM di infarto occipitale bilaterale, causa della sindrome di Anton
Ricardo BM, et al. Anton syndrome after subarachnoid hemorrhage and delayed cerebral ischemia: A case report. Cereb Circ Cogn Behav. 2021. Figure 1. PMCID: PMC9616440. License: CC BY.
La TC mostra emorragia ed ematoma nel lobo frontale destro, mentre le sequenze FLAIR e T2-pesate della RM mostrano infarti emorragici bilaterali nei lobi occipitali. Corrisponde all’infarto occipitale bilaterale discusso nella sezione «2. Principali sintomi e segni clinici».
Negazione della perdita visiva: il paziente non riconosce la propria perdita della vista. Anche quando urta oggetti, attribuisce la causa a fattori esterni, come la scarsa illuminazione della stanza.
Confabulazione: descrizione dettagliata di persone o situazioni inesistenti. Quando si chiede una stretta di mano, allunga la mano nella direzione sbagliata.
Conservazione parziale della visione dei colori: la visione dei colori può essere preservata. A volte è possibile riconoscere oggetti in movimento ma non quelli fermi. Ciò è dovuto a una via sottocorticale che bypassa V1 verso l’area V5. 1)
Reperti clinici (reperti riscontrati dal medico durante l’esame)
Acuità visiva: perdita completa (NLP: nessuna percezione della luce), ma il paziente non se ne accorge.
Riflesso pupillare: normale. Le lesioni del lobo occipitale si trovano posteriormente al corpo genicolato laterale, quindi non influenzano la via pupillare. La via afferente del riflesso fotomotore si dirige al pretetto mesencefalico prima del corpo genicolato laterale.
Esame del fondo oculare: normale. Non si riscontrano anomalie organiche intraoculari. 1)
Riflesso di ammiccamento alla minaccia: negativo.
Movimenti oculari: segue normalmente i comandi verbali, ma è incapace di inseguimento visivo.
Fenomeno di Riddoch: incapacità di riconoscere oggetti statici ma capacità di riconoscere oggetti in movimento. Descritto da Riddoch nel 1917, è noto come una caratteristica della cecità corticale.
Blindsight: fenomeno in cui, nonostante l’incapacità di percepire consapevolmente uno stimolo visivo, si reagisce inconsciamente ad esso. Si ipotizza il coinvolgimento di vie visive alternative alla via LGB-V1 (V2, V3, V4, V5/MT, FST, LIP).
QPerché il riflesso pupillare è normale nonostante la cecità corticale?
A
La via afferente del riflesso pupillare alla luce si dirige al pretetto mesencefalico prima del corpo genicolato laterale (LGB) e non passa attraverso il lobo occipitale (corteccia visiva primaria). Pertanto, anche in presenza di un danno al lobo occipitale, il riflesso pupillare è preservato. Insieme alla normalità del fundus oculare, questo costituisce un reperto caratteristico della cecità corticale.
Di seguito sono elencate le principali cause e i fattori di rischio associati alla sindrome di Anton.
Cause vascolari
Infarto cerebrale bilaterale delle PCA: causa più frequente. L’infarto di entrambe le arterie cerebrali posteriori danneggia estesamente i lobi occipitali.
Post-traumatico o post-tumorale: può verificarsi dopo trauma cranico, tumore cerebrale o intervento chirurgico.
Chirurgia cardiaca e angiografia cerebrale: noti fattori di rischio iatrogeni.
Intossicazione da monossido di carbonio e PRES: può insorgere anche in caso di sindrome da leucoencefalopatia posteriore reversibile (PRES). Anche farmaci antitumorali come il cisplatino possono esserne causa.
Cause non vascolari
MELAS: encefalomiopatia mitocondriale. Sono stati riportati casi di insorgenza in pazienti con mutazione mt.3243A>G. 2)
Riacutizzazione di sclerosi multipla, encefalopatia ipertensiva in gravidanza, emorragia ostetrica: secondarie a diverse condizioni sistemiche.
Infezioni: encefalite da virus del Nilo occidentale (WNV), PML associata a HIV, ecc. 3)
Altro: adrenoleucodistrofia, vasculite del sistema nervoso centrale, ischemia associata a emorragia subaracnoidea, lesioni bilaterali delle radiazioni ottiche nella sindrome di Trousseau, ecc.
Caratteristica anatomica dell’arteria cerebrale posteriore (PCA): si dirama dall’arteria basilare, nella porzione prossimale (P1-P2) irrora le strutture profonde (talamo posteriore e mesencefalo), mentre nella porzione distale (P3-P4) irrora la corteccia occipitale. Il danno al segmento P4 è la causa principale del deficit del campo visivo. 1) La corteccia occipitale è lontana dal sistema vascolare centrale e ha una struttura vulnerabile all’ischemia.
QLa sindrome di Anton può verificarsi anche senza ictus?
A
Può manifestarsi anche per cause non vascolari come MELAS (encefalomiopatia mitocondriale) 2), SM (sclerosi multipla), encefalite da virus del Nilo occidentale 3), traumi, intossicazione da monossido di carbonio, ecc. In tutti i casi, la base patologica comune è la disfunzione bilaterale del lobo occipitale.
La diagnosi clinica si basa sulla combinazione di quattro elementi: anamnesi di confabulazione, evidenza clinica di perdita della vista, fundus oculare normale e conferma di lesione del lobo occipitale tramite imaging.
TC cranica (NCCT): utile per la valutazione d’urgenza. Consente di identificare l’infarto ischemico (area ipodensa). Ad esempio, si presenta come un infarto ischemico del lobo temporo-occipitale destro. 1)
RM cranica:
Nella fase iperacuta (entro 6 ore), la DWI è utile. La DWI può rilevare infarti acuti difficili da identificare con T1, T2 e FLAIR.
Le immagini FLAIR sono eccellenti per distinguere l’infarto cerebrale dal liquido cerebrospinale.
Si osservano aree iperintense nella corteccia occipitale e nella sostanza bianca sottocorticale in T2-FLAIR. 2)3)
Interpretazione dei reperti DWI: iperintensità DWI con ADC isointenso (senza riduzione) suggerisce alterazioni dovute a crisi epilettiche, da differenziare dall’ictus ischemico (che presenta riduzione dell’ADC). 2)
VEP (potenziali evocati visivi): Utile per confermare la cecità corticale completa. Può dimostrare oggettivamente l’assenza di risposta agli stimoli ed è utilizzato anche per la diagnosi differenziale con la simulazione.
V-EEG (videoelettroencefalogramma): Utile quando si sospettano crisi epilettiche in patologie come la MELAS. Permette di rilevare crisi di origine occipitale. 2)
Nessuna anomalia oculare o del riflesso pupillare (comune con la cecità corticale), necessaria valutazione psichiatrica
La diagnosi differenziale con il disturbo visivo psicogeno è particolarmente importante. La cecità corticale dovuta a danno bilaterale dei lobi occipitali non presenta anomalie oculari e il riflesso pupillare alla luce è conservato, per cui può essere facilmente scambiata per una malattia psichiatrica o simulazione.
Il trattamento della sindrome di Anton viene scelto in base alla causa presunta del danno al lobo occipitale. Il trattamento della causa di base è fondamentale.
Da SM: terapia con steroidi in bolo (metilprednisolone EV) + plasmaferesi. Sono stati riportati casi di recupero graduale nell’arco di 2 anni, prima del insight e poi della vista.
Da MELAS: ottimizzazione dei farmaci antiepilettici (aggiustamento del dosaggio di lorazepam e levetiracetam, aggiunta di lacosamide) + terapia sostitutiva (L-arginina, levocarnitina, vitamine C, B1, B2, B12). 2)
Da encefalite da WNV: sono stati riportati casi in cui metilprednisolone 1000 mg/die per 7 giorni è stato tentato senza risposta; non esiste una terapia consolidata. 3)
QQuanto tempo dall'esordio è importante per il trattamento?
A
In caso di origine da ictus, entro 4,5 ore dall’esordio è indicata la tPA (terapia trombolitica). 1) Oltre questo tempo, la prevenzione delle recidive e la riabilitazione diventano il cardine del trattamento. Poiché maggiore è il tempo a disposizione, più ampia è la possibilità di salvare i neuroni, è importante recarsi immediatamente al pronto soccorso alla comparsa dei sintomi.
6. Fisiopatologia e meccanismi dettagliati di insorgenza
Un danno bilaterale esteso al lobo occipitale (corteccia visiva primaria V1) provoca un’emianopsia omonima bilaterale, portando infine alla cecità corticale. La corteccia occipitale è lontana dal sistema vascolare centrale ed è vulnerabile all’ischemia. Un infarto delle porzioni distali dell’arteria cerebrale posteriore (P3-P4) interrompe il flusso sanguigno al lobo occipitale. 1)
L’elaborazione delle informazioni visive comprende la via ventrale (via del “cosa”: area V4, riconoscimento di forma e colore) e la via dorsale (via del “dove”: area V5, posizione spaziale e movimento). Alcune fibre sottocorticali bypassano V1 e si collegano direttamente a V4 e V5, quindi anche se V1 è ampiamente danneggiato, queste vie possono funzionare, consentendo la conservazione della visione dei colori e il riconoscimento del movimento. 1) Questa è la base neurologica del fenomeno di Riddoch (vedere oggetti in movimento).
Per quanto riguarda il meccanismo della visione cieca (blindsight), studi su macachi hanno riportato proiezioni dirette dal LGBd alle aree V2, V3, V4, V5/MT, FST e LIP, e questa via potrebbe spiegare le risposte visive inconsce.
Meccanismo di insorgenza dell’anosognosia (confabulazione)
Esistono diverse ipotesi riguardo al meccanismo dell’anosognosia.
Ipotesi
Contenuto
Evidenza
Danno simultaneo della corteccia visiva e associativa
La corteccia visiva primaria e la corteccia associativa visiva sono danneggiate contemporaneamente, portando a una mancanza di consapevolezza del proprio stato
Quadro clinico di lesioni occipitali estese
Sindrome da disconnessione
Le lesioni della sostanza bianca parietale interrompono la connessione tra la corteccia visiva e altre aree
Insorgenza in casi con lesioni della sostanza bianca
Anomalia di connessione con il centro del linguaggio
La connessione tra la corteccia visiva danneggiata e le aree linguistiche funzionanti è interrotta, e le aree linguistiche generano risposte confabulatorie senza input visivo
Il contenuto della confabulazione include dettagli visivi
Attualmente, la teoria dell‘“anomalia di connessione con il centro del linguaggio” è la più supportata. Si ritiene che l’interruzione del feedback dalla corteccia visiva danneggiata alle aree linguistiche porti queste ultime a generare un falso resoconto di “vedere”.
QPerché i pazienti affermano di vedere quando in realtà non vedono?
A
L’ipotesi più supportata è quella dell‘“anomalia di connessione con il centro del linguaggio”. Il danno al lobo occipitale interrompe il circuito di feedback dalla corteccia visiva alle aree linguistiche, che quindi generano una risposta confabulatoria di “vedere” senza input visivo. Non si tratta di una bugia intenzionale, ma di un fenomeno neurologico dovuto a un danno dei circuiti cerebrali.
Meccanismo nella MELAS: Mitocondri anomali si accumulano nelle cellule endoteliali e muscolari lisce delle piccole arteriole, causando proliferazione capillare. Le crisi epilettiche provocano un rapido esaurimento energetico nell’unità neurovascolare, portando a una patologia simile alla paralisi di Todd. L’ipotesi di Fryer propone che “le crisi epilettiche siano il fattore scatenante degli episodi simili a ictus”. 2)
Meccanismo di passaggio della barriera ematoencefalica (BBB) nell’encefalite da WNV: sono ipotizzate tre vie: trasporto cellulare passivo, trasporto assonale e rottura della BBB indotta dall’infiammazione. 3)
7. Ricerche recenti e prospettive future (rapporti in fase di studio)
Ziaul et al. (2024) in un case report hanno riportato che i pazienti con COVID-19 presentano un rischio di ictus ischemico aumentato di 3,6 volte e che anche il COVID-19 lieve aumenta il rischio di ictus a circa l’1%. 1) Si ritiene che l’elevata risposta infiammatoria, lo stato di ipercoagulabilità e la gravità medica predispongano a eventi tromboembolici. Anche negli ictus della circolazione posteriore, inclusa l’arteria cerebrale posteriore (PCA), il ritardo nella presentazione è stato un problema durante la pandemia.
Reversibilità delle alterazioni DWI indotte da crisi epilettiche
Ewida et al. (2021) hanno riportato un caso di sindrome di Anton-Babinski associata a MELAS con reperti MRI caratteristici: iperintensità DWI e segnale ADC iso-intenso (incoerente con alterazioni ischemiche). 2) Questo reperto differisce dalle alterazioni DWI dell’ictus ischemico (accompagnate da riduzione ADC) ed è interpretato come una combinazione di disturbo energetico reversibile e alterazioni emodinamiche dovute a crisi epilettiche. La diagnosi differenziale dei reperti DWI fornisce importanti indicazioni per la decisione terapeutica nella MELAS.
Futuri candidati terapeutici per l’encefalite da WNV
Srichawla (2022) ha riportato che l’interferone alfa e le immunoglobuline purificate contenenti anticorpi anti-WNV potrebbero essere futuri candidati terapeutici per l’infezione neuroinvasiva da WNV. 3) Attualmente, la risposta ai farmaci esistenti, inclusa la metilprednisolone, è scarsa e non esiste una terapia consolidata. Oltre l’80% delle infezioni da WNV è asintomatico, ma meno del 5% progredisce verso la forma neuroinvasiva, rendendo urgente lo sviluppo di terapie.
Ziaul YH, Mittal J, Afroze T, et al. Anton-Babinski Syndrome: A Visual Anosognosia. Cureus. 2024;16(3):e55679.
Ewida A, Ahmed R, Luo A, et al. Mitochondrial Myopathy, Encephalopathy, Lactic acidosis and Stroke-Like Episodes Syndrome Presenting With Anton-Babinski Syndrome and Concurrent Occipital Lobe Seizures. Cureus. 2021;13(1):e12908.
Srichawla BS. Neuroinvasive West Nile Virus (WNV) Encephalitis With Anton Syndrome: Epidemiology and Pathophysiology Review. Cureus. 2022;14(6):e26264.
Copia il testo dell'articolo e incollalo nell'assistente IA che preferisci.
Articolo copiato negli appunti
Apri un assistente IA qui sotto e incolla il testo copiato nella chat.