Das Anton-Syndrom ist ein Zustand der visuellen Anosognosie, bei dem Patienten mit kortikaler Blindheit ihren eigenen Sehverlust nicht erkennen und über nicht vorhandene visuelle Erfahrungen berichten. Obwohl sie blind sind, neigen sie zu Konfabulationen, indem sie detailliert beschreiben, was sie angeblich sehen.
Die Erkrankung ist nach dem österreichischen Neurologen Gabriel Anton benannt. Anton berichtete über einen 69-jährigen Patienten mit bilateralen Temporallappenläsionen, der eine erworbene Anosognosie und Taubheit aufwies. Der Begriff „Anosognosie“ wurde von Joseph Babinski geprägt, weshalb das Syndrom auch als „Anton-Babinski-Syndrom“ bezeichnet wird. Die erste Beschreibung einer visuellen Anosognosie geht auf die römische Sklavin Harpaste zurück. Harpaste leugnete ihre Blindheit und behauptete, der Raum sei dunkel.
Abgrenzung zum Charles-Bonnet-Syndrom: Das Charles-Bonnet-Syndrom tritt bei Patienten mit Sehbehinderung auf, die visuelle Halluzinationen erleben, jedoch ihre Sehbehinderung erkennen – ein grundlegender Unterschied zum Anton-Syndrom.
Epidemiologie: Das mediane Alter beträgt 55 Jahre (Spanne 6–96 Jahre), es gibt keine Geschlechterunterschiede. Am häufigsten tritt es als Folge eines zerebrovaskulären Ereignisses (CVA) auf, vor allem bei älteren Menschen mit mehreren vaskulären Risikofaktoren. Ein Schlaganfall der hinteren Hirnarterie (PCA) macht etwa 5–10 % aller Schlaganfälle aus. 1) Zwischen 1965 und 2016 wurden nur 28 Fälle des Anton-Babinski-Syndroms berichtet, was es zu einer äußerst seltenen Erkrankung macht. 2)
QWas ist der Unterschied zwischen Anton-Syndrom und Charles-Bonnet-Syndrom?
A
Das Anton-Syndrom ist eine Erkrankung, bei der Patienten mit kortikaler Blindheit ihre eigene Blindheit leugnen und konfabulieren; es fehlt die Einsicht. Beim Charles-Bonnet-Syndrom hingegen haben sehbehinderte Patienten visuelle Halluzinationen, behalten aber die Einsicht in ihre Sehbehinderung. Beide haben eine Sehstörung als Hintergrund, unterscheiden sich jedoch grundlegend im Vorhandensein oder Fehlen von Anosognosie.
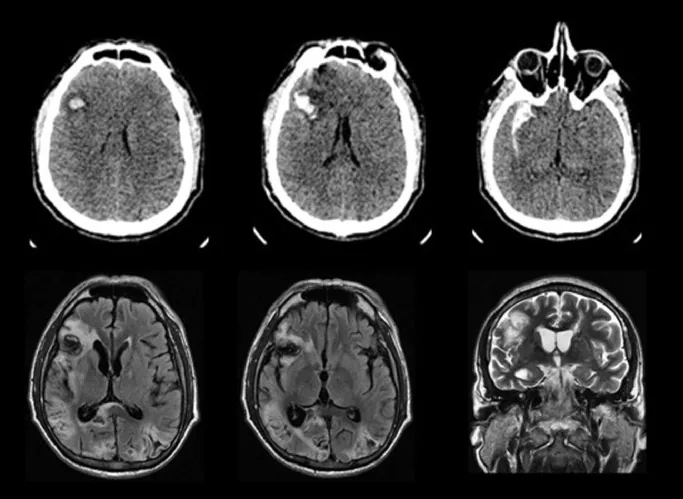
CT- und MRT-Bilder eines bilateralen Hinterhauptlappeninfarkts, der das Anton-Syndrom verursacht
Ricardo BM, et al. Anton syndrome after subarachnoid hemorrhage and delayed cerebral ischemia: A case report. Cereb Circ Cogn Behav. 2021. Figure 1. PMCID: PMC9616440. License: CC BY.
Im CT sind eine Blutung und ein Hämatom im rechten Frontallappen zu sehen, während die FLAIR- und T2-gewichteten MRT-Aufnahmen hämorrhagische Infarkte in beiden Hinterhauptlappen zeigen. Dies entspricht dem bilateralen Hinterhauptlappeninfarkt, der im Abschnitt „2. Hauptsymptome und klinische Befunde“ behandelt wird.
Leugnen des Sehverlusts: Der Patient erkennt seinen eigenen Sehverlust nicht. Wenn er gegen Gegenstände stößt, führt er dies auf äußere Faktoren zurück, z. B. darauf, dass der Raum dunkel sei.
Konfabulation: Detaillierte Beschreibung nicht existierender Personen oder Situationen. Beim Händeschütteln wird die Hand in die falsche Richtung ausgestreckt.
Teilerhalt des Farbsehens: Das Farbsehen kann erhalten bleiben. Bewegte Objekte werden erkannt, aber das Erkennen von Stillleben kann schwierig sein. Dies ist auf subkortikale Faserbahnen zurückzuführen, die das V5-Areal unter Umgehung von V1 erreichen. 1)
Klinische Befunde (vom Arzt bei der Untersuchung festgestellte Befunde)
Sehschärfe: Vollständiger Verlust (NLP: no light perception), aber der Patient bemerkt es nicht.
Pupillenreflex: Normal. Da die Läsion im Okzipitallappen hinter dem Corpus geniculatum laterale liegt, wird die Pupillenbahn nicht beeinträchtigt. Die afferente Bahn des Lichtreflexes verläuft vor dem Corpus geniculatum laterale zur Area praetectalis des Mittelhirns.
Funduskopie: Normal. Es liegen keine organischen Auffälligkeiten im Augapfel vor. 1)
Drohblinzelreflex: Negativ.
Augenbewegungen: Auf verbale Anweisungen wird normal reagiert, aber visuelles Folgen ist nicht möglich.
Kornealreflex: normal (unabhängig von kortikaler Eingabe).
Riddoch-Phänomen: Unfähigkeit, stationäre Objekte zu erkennen, aber bewegte Objekte werden wahrgenommen. 1917 von Riddoch beschrieben, bekannt als ein Merkmal der kortikalen Blindheit.
Blindsight: Phänomen, bei dem visuelle Reize nicht bewusst wahrgenommen werden, aber unbewusste Reaktionen auf die Reize erfolgen. Beteiligung visueller Bahnen außerhalb des LGB-V1-Wegs (V2, V3, V4, V5/MT, FST, LIP) wird vermutet.
QWarum ist der Pupillenreflex bei kortikaler Blindheit normal?
A
Der afferente Schenkel des Lichtreflexes zieht vor dem Corpus geniculatum laterale (CGL) zum prätektalen Bereich des Mittelhirns und umgeht den Okzipitallappen (primären visuellen Kortex). Daher bleibt der Lichtreflex auch bei Läsionen des Okzipitallappens erhalten. Zusammen mit einem normalen Fundusbefund ist dies ein charakteristischer Befund der kortikalen Blindheit.
Kohlenmonoxidvergiftung/PRES: Auch bei toxischen oder reversiblen posterioren Leukenzephalopathie-Syndrom (PRES) möglich. Zytostatika wie Cisplatin können ebenfalls ursächlich sein.
Nicht-vaskuläre Ursachen
MELAS: Mitochondriale Enzephalomyopathie. Berichte über Auftreten bei Patienten mit mt.3243A>G-Mutation. 2)
MS-Schub, Schwangerschaftshypertonie, geburtshilfliche Blutungen: Sekundär zu verschiedenen systemischen Erkrankungen.
Sonstige: Adrenoleukodystrophie, ZNS-Vaskulitis, Ischämie nach Subarachnoidalblutung, bilaterale Sehstrahlungsläsionen beim Trousseau-Syndrom usw.
Anatomisch gesehen zweigt die hintere Hirnarterie (PCA) von der Basilararterie ab; der proximale Abschnitt (P1–P2) versorgt tiefe Strukturen (hinterer Thalamus, Mittelhirn), der distale Abschnitt (P3–P4) die okzipitale Kortikalis. Eine Schädigung des P4-Segments ist die Hauptursache für Gesichtsfeldausfälle. 1) Die okzipitale Kortikalis ist weit vom zentralen Gefäßsystem entfernt und strukturell anfällig für Ischämie.
QKann das Anton-Syndrom auch ohne Schlaganfall auftreten?
A
MELAS (Mitochondriale Enzephalomyopathie) 2), MS (Multiple Sklerose), West-Nil-Virus-Enzephalitis 3), Trauma, Kohlenmonoxidvergiftung und andere nicht-vaskuläre Ursachen können ebenfalls zum Anton-Syndrom führen. Allen gemeinsam ist eine beidseitige Funktionsstörung des Okzipitallappens als pathophysiologische Grundlage.
Die klinische Diagnose erfolgt durch die Kombination von vier Punkten: „Anamnese von Konfabulationen + klinischer Nachweis von Sehverlust + normaler Augenhintergrundbefund + bildgebender Nachweis einer Okzipitallappenläsion“.
Kopf-CT (NCCT): Nützlich für die Notfallbeurteilung. Nachweis eines ischämischen Infarkts (Hypodensität). Beispielsweise als ischämischer Infarkt des rechten temporo-okzipitalen Kortex dargestellt. 1)
MRT des Kopfes:
In der hyperakuten Phase (innerhalb von 6 Stunden) ist die DWI nützlich. Auch akute Infarkte, die in T1-, T2- und FLAIR-Aufnahmen schwer zu erkennen sind, können mit DWI erfasst werden.
FLAIR-Aufnahmen eignen sich hervorragend zur Unterscheidung von Hirninfarkt und Liquor.
Nachweis von hyperintensen Arealen im okzipitalen Kortex und subkortikalen Marklager in T2-FLAIR. 2)3)
Interpretation der DWI-Befunde: DWI-Hyperintensität mit isointensem ADC (keine Erniedrigung) deutet auf epilepsiebedingte Veränderungen hin und wird vom ischämischen Schlaganfall (mit ADC-Erniedrigung) unterschieden. 2)
VEP (visuell evozierte Potenziale): Nützlich zur Bestätigung einer vollständigen kortikalen Blindheit. Kann das Fehlen einer Reaktion auf Reize objektiv nachweisen und wird auch zur Abgrenzung von Simulation verwendet.
V-EEG (Video-EEG): Nützlich bei Verdacht auf epileptische Anfälle, z. B. bei MELAS. Kann Anfälle okzipitalen Ursprungs erfassen. 2)
Keine Auffälligkeiten bei Augen- und Lichtreflex (gemeinsam mit Rindenblindheit), psychiatrische Beurteilung erforderlich
Die Abgrenzung zur psychogenen Sehstörung ist besonders wichtig. Die durch bilaterale Okzipitallappenschädigung verursachte Rindenblindheit weist keine Auffälligkeiten am Auge auf und der Pupillenlichtreflex bleibt erhalten, sodass sie leicht mit psychischen Erkrankungen oder Simulation verwechselt wird.
Intravenöse tPA (Gewebe-Plasminogen-Aktivator): Indikation innerhalb von 4,5 Stunden nach Symptombeginn. Thrombolyse verhindert die Ausdehnung des Infarktareals. 1)
Endovaskuläre Behandlung: In der hyperakuten Phase wird eine endovaskuläre Katheterbehandlung (Thrombektomie) in Betracht gezogen.
Bei mehr als 4,5 Stunden nach Symptombeginn: Fokus auf weitere Schlaganfallprävention und Rehabilitation. 1)
MS-bedingt: Steroid-Pulstherapie (IV Methylprednisolon) + Plasmaaustausch. Es wurden Fälle berichtet, bei denen sich die Einsicht und dann das Sehvermögen über 2 Jahre allmählich erholten.
MELAS-bedingt: Optimierung der Antiepileptika (Dosisanpassung von Lorazepam und Levetiracetam, Hinzufügen von Lacosamid) + Supplementierung (L-Arginin, Levocarnitin, Vitamin C, B1, B2, B12). 2)
WNV-Enzephalitis-bedingt: Methylprednisolon 1000 mg/Tag für 7 Tage wurde versucht, zeigte jedoch keine Wirkung; es gibt keine etablierte Behandlung. 3)
QWie viel Zeit nach Symptombeginn ist für die Behandlung entscheidend?
A
Bei Schlaganfall ist tPA (Thrombolyse) innerhalb von 4,5 Stunden nach Symptombeginn indiziert. 1) Danach stehen Rezidivprophylaxe und Rehabilitation im Vordergrund. Je früher die Behandlung erfolgt, desto mehr Nervenzellen können gerettet werden. Daher ist bei Auftreten von Symptomen eine sofortige Notaufnahme wichtig.
6. Pathophysiologie und detaillierter Entstehungsmechanismus
Eine beidseitige ausgedehnte Schädigung des Okzipitallappens (primärer visueller Kortex V1) führt zu einer bilateralen homonymen Hemianopsie und schließlich zur kortikalen Blindheit. Der okzipitale Kortex ist weit vom zentralen Gefäßsystem entfernt und anfällig für Ischämie. Ein Infarkt der distalen Abschnitte der hinteren Hirnarterie (P3–P4) unterbricht die Durchblutung des Okzipitallappens. 1)
Die visuelle Informationsverarbeitung umfasst den ventralen Pfad („Was“-Pfad: V4, Form- und Farberkennung) und den dorsalen Pfad („Wo“-Pfad: V5, räumliche Position und Bewegungserkennung). Einige subkortikale Fasern umgehen V1 und verbinden sich direkt mit V4 und V5. Daher können bei ausgedehnter V1-Schädigung diese Pfade funktionieren und Farberkennung oder Bewegungswahrnehmung ermöglichen. 1) Dies ist die neurologische Grundlage des Riddoch-Phänomens (Sehen von Bewegung).
Der Mechanismus des Blindsight wurde in Studien an Makaken beschrieben, die eine direkte Projektion vom LGBd zu V2, V3, V4, V5/MT, FST und LIP zeigen. Dieser Pfad könnte unbewusste visuelle Reaktionen erklären.
Entstehungsmechanismus der Anosognosie (Konfabulation)
Zur Pathogenese der Anosognosie (Konfabulation) existieren mehrere Hypothesen.
Hypothese
Inhalt
Evidenz
Gleichzeitige Schädigung des visuellen Kortex und Assoziationskortex
Primärer visueller Kortex und visueller Assoziationskortex sind gleichzeitig geschädigt, was zu einem Mangel an Einsicht in den eigenen Zustand führt.
Klinisches Bild ausgedehnter okzipitaler Läsionen
Diskonnektionssyndrom
Läsionen der parietalen weißen Substanz unterbrechen die Verbindung zwischen visuellem Kortex und anderen Regionen.
Auftreten bei Fällen mit Läsionen der weißen Substanz
Verbindungsstörung mit dem Sprachzentrum
Die Verbindung zwischen dem geschädigten visuellen Kortex und dem funktionierenden Sprachareal ist unterbrochen, sodass das Sprachareal ohne visuellen Input konfabulatorische Antworten generiert
Der Inhalt der Konfabulationen enthält visuelle Details
Derzeit wird die Theorie der „Verbindungsstörung mit dem Sprachzentrum“ am meisten unterstützt. Es wird angenommen, dass durch den Ausfall des Feedbacks vom geschädigten visuellen Kortex zum Sprachareal dieses falsche Berichte darüber generiert, dass etwas „gesehen“ wird.
QWarum behaupten Patienten, etwas zu sehen, obwohl sie es nicht sehen?
A
Die am meisten unterstützte Hypothese ist die Theorie der „Verbindungsstörung mit dem Sprachzentrum“. Durch eine Schädigung des Okzipitallappens wird die Feedback-Schleife vom visuellen Kortex zum Sprachareal unterbrochen, und das Sprachareal generiert ohne visuellen Input konfabulatorische Antworten, dass etwas „gesehen“ wird. Dies ist keine absichtliche Lüge, sondern ein neurologisches Phänomen, das auf einer Schädigung der Hirnbahnen beruht.
Krankheitsspezifische Pathophysiologie nach Ursache
Mechanismus bei MELAS: In den Endothel- und glatten Muskelzellen kleiner Arteriolen akkumulieren abnormale Mitochondrien, was zu einer Kapillarproliferation führt. Epileptische Anfälle verursachen eine rasche Energieverarmung der neurovaskulären Einheit, was zu einem Zustand ähnlich der Todd-Parese führt. Die Fryer-Hypothese besagt, dass „epileptische Anfälle den Auslöser für stroke-like Episoden darstellen“. 2)
Mechanismus der Blut-Hirn-Schranken-Passage bei WNV-Enzephalitis: Es werden drei Wege angenommen: passiver Zelltransport, axonaler Transport und entzündungsinduzierte BBB-Zerstörung. 3)
7. Aktuelle Forschung und zukünftige Perspektiven (Berichte aus der Forschungsphase)
Ziaul et al. (2024) berichteten in einem Fallbericht, dass COVID-19-Patienten ein 3,6-fach erhöhtes Risiko für ischämischen Schlaganfall haben und dass selbst bei mildem COVID-19 das Schlaganfallrisiko auf etwa 1 % ansteigt. 1) Eine hohe Entzündungsreaktion, ein hyperkoagulierbarer Zustand und die medizinische Schwere gelten als prädisponierende Faktoren für Thromboembolien. Auch bei Schlaganfällen des hinteren Kreislaufs, einschließlich der PCA, war die verzögerte Vorstellung während der Pandemie ein Problem.
Ewida et al. (2021) berichteten über einen Fall von MELAS-assoziiertem Anton-Babinski-Syndrom mit charakteristischen MRT-Befunden: DWI-Hyperintensität und ADC-Isosignal (nicht vereinbar mit ischämischen Veränderungen). 2) Dieser Befund unterscheidet sich von DWI-Veränderungen bei ischämischem Schlaganfall (mit ADC-Abnahme) und wird als Kombination aus reversibler Energie-Stoffwechselstörung und hämodynamischen Veränderungen infolge epileptischer Anfälle interpretiert. Die Differenzierung der DWI-Befunde liefert wichtige Hinweise für die Therapieentscheidung bei MELAS.
Zukünftige Behandlungskandidaten für WNV-Enzephalitis
Srichawla (2022) berichtete, dass Interferon-alpha und gereinigte Immunglobulinpräparate mit WNV-Antikörpern zukünftige Behandlungskandidaten für neuroinvasive WNV-Infektionen sein könnten. 3) Derzeit ist die Reaktion auf bestehende Medikamente einschließlich Methylprednisolon gering, und es gibt keine etablierte Therapie. Über 80 % der WNV-Infektionen verlaufen asymptomatisch, aber weniger als 5 % entwickeln sich zu einer neuroinvasiven Form, sodass die Etablierung einer Behandlung dringend erforderlich ist.
Ziaul YH, Mittal J, Afroze T, et al. Anton-Babinski Syndrome: A Visual Anosognosia. Cureus. 2024;16(3):e55679.
Ewida A, Ahmed R, Luo A, et al. Mitochondrial Myopathy, Encephalopathy, Lactic acidosis and Stroke-Like Episodes Syndrome Presenting With Anton-Babinski Syndrome and Concurrent Occipital Lobe Seizures. Cureus. 2021;13(1):e12908.
Srichawla BS. Neuroinvasive West Nile Virus (WNV) Encephalitis With Anton Syndrome: Epidemiology and Pathophysiology Review. Cureus. 2022;14(6):e26264.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.