Das Merkelzellkarzinom ist ein seltener neuroendokriner Tumor, der von Merkelzellen in der Basalschicht der Epidermis ausgeht. Merkelzellen, 1875 von Friedrich Merkel entdeckt, sind sensorische Zellen, die an der Wahrnehmung von leichtem Berührungsreiz und der Unterscheidung von Form und Textur beteiligt sind.
Die jährliche Inzidenz beträgt 0,23 Fälle pro 100.000 Personen. Etwa 43–50 % aller Fälle treten im Kopf-Hals-Bereich auf, davon 5–10 % an den Augenlidern1). Von 2000 bis 2013 stieg die Zahl der Merkelzellkarzinom-Fälle um 95 %, was deutlich schneller ist als bei allen soliden Tumoren (15 % Anstieg) oder Melanomen (56 % Anstieg)1). Hintergrund sind die alternde Bevölkerung, die Zunahme immunsupprimierter Patienten und verbesserte Diagnosetechniken.
Am Augenlid tritt es typischerweise an der Wimpernlinie auf, rot, mit gespannter und glänzender Haut. Aufgrund des schnellen Wachstums und der Neigung zu lymphogener Metastasierung sind eine schnelle Diagnose und Behandlung erforderlich.
QWie selten ist das Merkelzellkarzinom?
A
Die jährliche Inzidenz beträgt 0,23 Fälle pro 100.000 Personen, was es zu einem seltenen Hautmalignom macht. In den letzten Jahren hat es jedoch rapide zugenommen, mit einem Anstieg der Fallzahlen um 95 % zwischen 2000 und 20131).
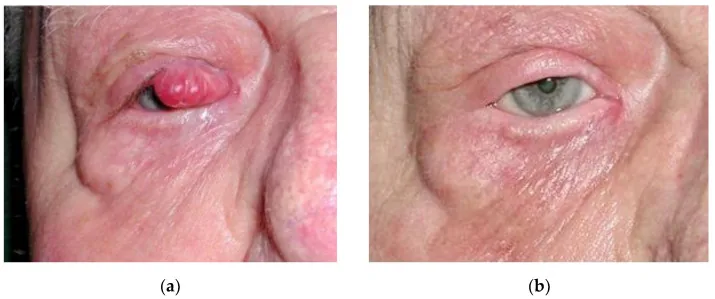
Boileau M, et al. An Effective Primary Treatment Using Radiotherapy in Patients with Eyelid Merkel Cell Carcinoma. Curr Oncol. 2023. Figure 2. PMCID: PMC10377768. License: CC BY.
4 Jahre nach kurativer Strahlentherapie eines 20 mm Merkelzellkarzinoms des rechten Augenlids, (a) vor Behandlung, (b) nach Behandlung. Entspricht dem Augenlid-Tumor, der im Abschnitt „2. Hauptsymptome und klinische Befunde“ behandelt wird.
Das Merkelzellkarzinom ist in der Regel schmerzlos und weist anfangs wenige subjektive Symptome auf. Es wird oft als schnell wachsender Hautknoten bemerkt.
Klinische Befunde (vom Arzt bei der Untersuchung festgestellte Befunde)
Das Merkelzellkarzinom erscheint meist als einzelner, schmerzloser Knoten von „violetter“ Farbe. Zur Diagnosehilfe wurden die AEIOU-Kriterien vorgeschlagen.
Kriterium
Inhalt
Asymptomatisch
Asymptomatisch (keine Druckempfindlichkeit)
Expandiert schnell
Schnelle Ausdehnung
Immunsuppression
Immunsupprimierter Zustand
Older than 50 (älter als 50)
Alter über 50 Jahre
UV-exponierte Stelle
UV-exponierte Stelle
89 % der Patienten mit Merkelzellkarzinom erfüllen drei oder mehr Kriterien, 52 % erfüllen vier oder mehr.
Periokulär tritt es am häufigsten in der Nähe der Wimpernlinie des Oberlids auf. Es bildet einen roten, kuppelförmigen Knoten mit erweiterten Tumorgefäßen. Charakteristisch sind teilweiser oder vollständiger Wimpernverlust (Madarosis) und oberflächliche Teleangiektasien.
Zum Zeitpunkt der Diagnose weisen bis zu 37 % der Patienten Lymphknotenmetastasen und 6–12 % Fernmetastasen auf. Die Palpation der präaurikulären, submandibulären und zervikalen Lymphknoten ist unerlässlich.
Merkelzell-Polyomavirus : etwa 80 % der Merkelzellkarzinome sind positiv (USA). In Japan etwa 90 % 1). Das virale Genom wird in das Wirtschromosom integriert, und das große T-Antigen wird mutiert und verkürzt, um ein Onkoprotein zu werden.
UV-Exposition : Merkelzell-Polyomavirus-negative Merkelzellkarzinome weisen häufig UV-induzierte Mutationen auf 1).
Immunsuppression : Organtransplantationsempfänger, chronische lymphatische Leukämie, HIV usw.
Hohes Alter : 50 Jahre und älter. Die Inzidenz steigt insbesondere nach dem 70. Lebensjahr 1).
Helle Haut : häufiger bei Weißen.
Viruspositives Merkelzellkarzinom
Merkelzell-Polyomavirus-assoziiert : etwa 80 % der Gesamtzahl. Virale Onkoproteine inaktivieren RB1 und p53. Hohe Immunogenität, Grundlage für T-Zell-gerichtete Therapien 1).
Zellulärer Ursprung : mehrere Theorien, darunter dermale Fibroblasten, Pro/Prä-B-Lymphozyten, epidermale Vorläuferzellen 1).
Virusnegatives Merkelzellkarzinom
UV-Mutations-assoziiert : Hohe Mutationslast. Expimiert UV-induzierte DNA-Neo-Antigene und ist hoch immunogen1).
Zellulärer Ursprung : Entsteht aus Keratinozyten/epidermalen Vorläuferzellen mit vielen UV-Mutationen. Es gibt Berichte über Kollisionstumoren mit Plattenepithelkarzinom1).
QFührt eine Infektion mit dem Merkelzell-Polyomavirus zum Merkelzellkarzinom?
A
Das Merkelzell-Polyomavirus ist ein nahezu allgegenwärtiges Virus, das auch in normaler Haut vorkommt, aber die Entwicklung eines Merkelzellkarzinoms ist äußerst selten und tritt bei etwa 1 von 3000 Menschen im Laufe des Lebens auf1). Zwei Ereignisse müssen in derselben Zelle stattfinden: die Integration des viralen Genoms in den Wirt und die Mutation des großen T-Antigens.
Die Tumorzellen haben ein spärliches Zytoplasma, einen großen runden Kern mit fein verteiltem Chromatin und zahlreiche Mitosefiguren. Sie bilden große Nester unter der Epidermis. Sie ähneln einem Lymphom, werden aber durch Immunhistochemie unterschieden.
Die punktförmige (perinukleäre Punktmuster) Positivität von Zytokeratin 20 ist das charakteristischste Merkmal des Merkelzellkarzinoms und zeigt eine Koexpression mit Pan-Zytokeratin2). Zytokeratin 20 wird bei Merkelzellkarzinom als zu 100 % positiv berichtet. Elektronenmikroskopisch finden sich im Zytoplasma neuroendokrine Granula mit elektronendichtem Kern.
Auch bei Patienten ohne klinische Lymphknotenmetastasen identifiziert die Sentinel-Lymphknotenbiopsie in einem Drittel der Fälle Mikrometastasen1). Patienten mit positiver Sentinel-Lymphknotenbiopsie haben ein etwa dreifach erhöhtes Rezidivrisiko im Vergleich zu negativen Patienten (3-Jahres-Rezidivrate: 60 % vs. 20 %), was für die Festlegung des Überwachungsplans wichtig ist1).
Die Basisbildgebung mittels FDG-PET/CT führt bei etwa jeder sechsten Person (16,8 %) zu einem Upstaging1). Die Häufigkeit okkulter Metastasen ist im Vergleich zum Melanom (<1 %) deutlich höher.
Metastasiertes kleinzelliges Lungenkarzinom: morphologisch am schwierigsten zu unterscheiden
QWie unterscheidet man das Merkelzellkarzinom vom kleinzelligen Lungenkarzinom?
A
Die Immunhistochemie ist der Schlüssel. Eine perinukleäre Punktmuster-Positivität für Zytokeratin 20 ist spezifisch für das Merkelzellkarzinom, während das kleinzellige Lungenkarzinom Zytokeratin 20-negativ und TTF-1-positiv ist2).
Grundlage der Behandlung ist die chirurgische Exzision und die pathologische Lymphknotenbeurteilung. An anderen Körperstellen werden Exzisionsränder von 1–2 cm empfohlen, aber im Augenbereich sind konservative Ränder von etwa 5 mm akzeptabel2). Die Überprüfung der Exzisionsränder erfolgt mittels Mohs-mikrografischer Chirurgie oder Gefrierschnitten.
Bei Fällen mit Adhäsion und Infiltration der Lidhaut und des Tarsus ist eine Volltexzision des Augenlids und eine Rekonstruktion analog zum Talgdrüsenkarzinom erforderlich. Nach Bestätigung negativer Ränder können 2–3 Zyklen Gefrieren und Auftauen (freeze and thaw) auf die Exzisionsfläche aufgetragen werden. Bei Patienten, die eine radikale Exzision nicht tolerieren, wird eine Strahlentherapie gewählt.
Indikationen: Die NCCN (National Comprehensive Cancer Network)-Leitlinien empfehlen eine adjuvante Strahlentherapie innerhalb von 4–6 Wochen nach chirurgischer Exzision für alle Stadien. Sie reduziert das 5-Jahres-Lokalrezidivrisiko erheblich.
Dosis: Üblicherweise 50–66 Gy2). Im Augenbereich ist auf die Auswirkungen auf die Augenstrukturen zu achten.
Radikale Strahlentherapie
Indikationen: Erstlinientherapie für nicht operationsfähige Patienten. Das Merkelzellkarzinom ist sehr strahlenempfindlich und kann manchmal allein mit Strahlentherapie behandelt werden.
Einzeitige Bestrahlung: Ansprechrate von über 94 % mit einer Einzeldosis von 8 Gy. Alternative Option für ältere/gebrechliche Patienten1).
Die konventionelle Chemotherapie mit Platin und Etoposid hat eine hohe initiale Ansprechrate, aber die Ansprechdauer ist kurz (bei den meisten Progression innerhalb von 90 Tagen) und eine Verbesserung des Überlebens wurde nicht gezeigt1).
Immun-Checkpoint-Inhibitoren sind zur Standard-Systemtherapie des fortgeschrittenen Merkelzellkarzinoms geworden:
Avelumab (Anti-PD-L1-Antikörper): 2017 von der FDA zugelassen. In der JAVELIN-Merkel-200-Studie Ansprechrate 33 %, vollständiges Ansprechen 11 %, 2-Jahres-progressionsfreies Überleben 26 %, Gesamtüberleben 36 %2). In vielen Regionen einschließlich Japan zugelassen.
Pembrolizumab (Anti-PD-1-Antikörper): 2018 von der FDA zugelassen. Ansprechrate in der Erstlinientherapie 55–62 %1).
Zeigt unabhängig vom Infektionsstatus mit Merkelzell-Polyomavirus oder PD-L1-Expression ähnliche Ansprechraten1).
Nach Chemotherapie in der Zweitlinie oder später sinkt die Ansprechrate auf etwa 30 %1).
QWie wirksam sind Immun-Checkpoint-Inhibitoren beim Merkelzellkarzinom?
A
Die Ansprechrate in der Erstlinientherapie beträgt 55–62 % und übertrifft damit die Chemotherapie deutlich1). Im Gegensatz zur Chemotherapie ist das Ansprechen oft anhaltend (mehrere Jahre). Eine Wirkung ist sowohl bei Virus-positiven als auch -negativen Fällen zu erwarten.
6. Pathophysiologie und detaillierte Entstehungsmechanismen
Es gibt zwei Hauptwege bei der Entstehung des Merkelzellkarzinoms.
Beim viruspositiven Merkelzellkarzinom inaktivieren die Onkoproteine des Merkelzell-Polyomavirus (verkürztes großes T-Antigen und kleines T-Antigen) RB1 und p53 und aktivieren die Myc-Signalgebung 1). Beim virusnegativen Merkelzellkarzinom führen UV-induzierte Mutationen direkt zu einer Fehlregulation dieser Signalwege. Bei beiden Typen treibt der „PARCB“-Faktor (Anomalien von p53, Akt1, RB1, c-Myc, Bcl2) die neuroendokrine Differenzierung voran 1).
Das Merkelzellkarzinom neigt zur diskontinuierlichen Ausbreitung, und lokale Rezidive können auch bei pathologisch negativen Resektionsrändern auftreten 1). Sechs Faktoren, die das Rezidivrisiko erhöhen, wurden berichtet: 1) chronische T-Zell-Immunsuppression, 2) Tumordurchmesser > 1 cm, 3) lymphovaskuläre Invasion, 4) positive Sentinel-Lymphknotenbiopsie, 5) positive Resektionsränder, 6) Primärlokalisation im Kopf-Hals-Bereich 1).
Die Messung zirkulierender Antikörper gegen die Onkoproteine des Merkelzell-Polyomavirus ist als prognostischer Indikator nützlich. Seronegative Patienten haben ein etwa 42 % höheres Rezidivrisiko als seropositive Patienten 1). Bei seropositiven Patienten kann der Verlauf des Antikörpertiters zur Früherkennung von Rezidiven genutzt werden, und diese Methode ist in den NCCN-Leitlinien als Überwachungsmethode aufgeführt 1).
7. Aktuelle Forschung und Zukunftsaussichten (Berichte aus der Forschungsphase)
Die präoperative (neoadjuvante) Gabe von Immun-Checkpoint-Inhibitoren führt bei etwa 50 % der Patienten mit lokal fortgeschrittenem Merkelzellkarzinom zu einer raschen Tumorverkleinerung 1). Eine fallbezogene Abwägung ist erforderlich.
Herausforderungen bei immuntherapieresistentem Merkelzellkarzinom
Etwa die Hälfte der Patienten mit fortgeschrittenem Merkelzellkarzinom spricht nicht dauerhaft auf eine Anti-PD-1/PD-L1-Therapie an 1). Der Umgang mit primärer oder erworbener Resistenz ist der größte ungedeckte Bedarf, und zahlreiche klinische Studien laufen.
Die Analyse zirkulierender Tumor-DNA (ctDNA) wird als vielversprechendes neues Instrument zur Früherkennung von Rezidiven bei Patienten mit virusnegativem Merkelzellkarzinom entwickelt 1). Ein stadienabhängiger Rezidivrisiko-Rechner (merkelcell.org/recur) trägt ebenfalls zur Personalisierung der Überwachung bei.