単純型
眼症状のみ:視覚障害が主体で、全身異常を伴わない。
中等度以上の遠視:全身異常を伴わない例に多い。

レーバー先天黒内障(Leber congenital amaurosis; LCA)は、出生時〜乳児期に重度の視覚障害を来す先天性網膜ジストロフィの中で最も重篤な型である。小児視覚障害の主要な原因疾患であり、臨床像は多彩である。
1869年にドイツの眼科医テオドール・フォン・レーバー(Theodor Karl Gustav von Leber, 1840–1917)によって初めて報告された。なお、同じレーバーが1871年に報告したレーバー遺伝性視神経症(LHON)は20歳前後で発症するミトコンドリア疾患であり、LCAとは全く異なる疾患である。1957年に網膜電図(ERG)での波形消失がLCA診断の共通特徴として確認され、疾患名が確立した。
推定出生有病率は出生10万人あたり2〜3人(1/30,000〜1/81,000)である1)。全網膜ジストロフィの約5%を占め、盲学校に通う視覚障害児の約20%がLCAである1)。現在約27のLCA関連遺伝子が同定されており2)、症例の約70〜80%で原因遺伝子が特定される。遺伝形式は常染色体劣性遺伝が主であるが、優性遺伝やX連鎖性遺伝の報告もある。
通常生後6週頃に眼振や固視欠如で親が気づくことが多い3)。重度の視覚反応不良(固視・追視が全くできない)が認められた場合にLCAを疑い、網膜電図で確定診断する。
出生時〜生後早期から重度の視覚障害が存在する。
生後6週頃に眼振や固視欠如で親が気づくことが多い3)。
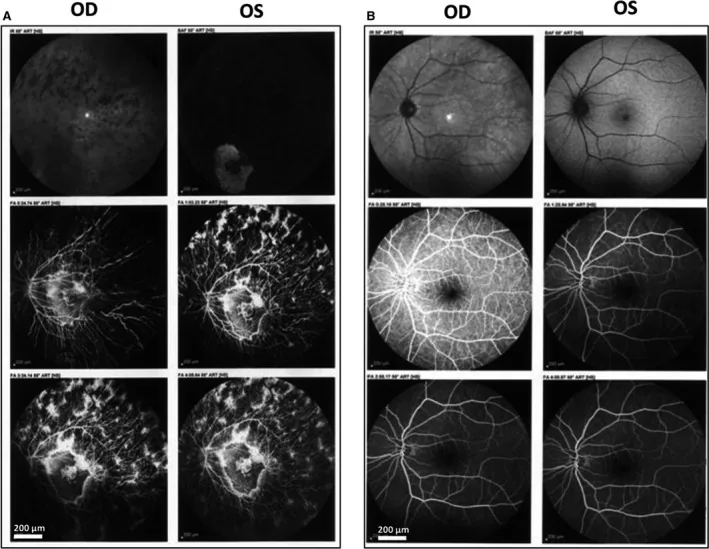
乳児期には眼底が正常に見えることも多いが、その後多彩な所見が出現する。眼底所見は正常眼底から定型的な網膜色素変性様眼底まで多種多様である。なお、進行した症例では眼底全体の色調が暗く、黄斑の輪状反射が消失する。
LCAは眼症状のみの単純型と全身疾患を合併する複雑型に分類される。
単純型
眼症状のみ:視覚障害が主体で、全身異常を伴わない。
中等度以上の遠視:全身異常を伴わない例に多い。
複雑型
中枢神経系異常:小脳虫部低形成、脳幹部奇形。
精神発達遅滞:知的障害を伴う例がある。
腎障害:嚢胞腎を合併することがある。
その他:難聴、骨格異常、肝障害、代謝異常、てんかん。
LCA患者に特徴的な行動で、目を指で突いたり押したりすることで網膜を機械的に刺激し、視覚を呼び起こそうとしていると考えられている。長期間の反復により眼窩脂肪の萎縮をきたし、眼球陥凹の原因となる。
LCAは遺伝性の網膜変性疾患群であり、大多数が常染色体劣性遺伝の形式をとる2)。まれにCRX・IMPDH1・OTX2の変異で常染色体優性遺伝を示す例がある2)。X連鎖性遺伝の報告も存在する。
現在LCA1〜LCA19の19型に加え、さらに8つの関連遺伝子が報告されている2)。原因遺伝子は光受容体形態形成、光情報伝達繊毛、視サイクル(ビジュアルサイクル)など、網膜の発達と機能に関わる複数の経路に関与している。
世界的に最も頻度の高い変異遺伝子とその割合は以下の通りである。
| 遺伝子 | 割合 | 関与する経路 |
|---|---|---|
| CEP290 | 約15% | 繊毛機能 |
| GUCY2D | 約12% | 光情報伝達(cGMP合成) |
| CRB1 | 約10% | 細胞極性維持 |
| RPE65 | 約8% | レチノイド代謝 |
日本人コホート(34家系)のNGS解析では検出率約56%であり、最頻変異遺伝子はCRB1・NMNAT1・RPGRIP1と報告されている1)。
常染色体劣性遺伝の場合、両親がともにキャリアであれば、次の子どもが罹患する確率は25%、キャリアとなる確率は50%、非罹患・非キャリアの確率は25%である。原因遺伝子が判明していれば出生前診断や着床前診断も選択肢となる。
LCAの診断は臨床的に行われ、網膜電図による確定診断と遺伝子検査による分子遺伝学的確認が必要である2)。
先天性の著しい視覚反応不良(固視・追視の欠如)を認めた場合にLCAを疑う。重度視力障害と強度遠視を伴う乳児では、分子遺伝学的検査によるLCAの検索が第一選択となる4)。
主な診断検査を以下に示す。
| 検査 | 所見・特徴 |
|---|---|
| 網膜電図(網膜電図) | 暗所・明所とも導出不能〜著明低下。必須 |
| OCT(光干渉断層計) | 網膜萎縮、黄斑陥凹、層構造の破壊 |
| FAF(眼底自発蛍光) | 亜型で異なる(後述) |
| 遺伝子検査(NGS等) | 確定診断・亜型同定に必要 |
ほとんどの型のLCAに対して実質的な治療法は確立されていない。現時点での管理は以下の通りである。
2017年、米国FDAは両アレル性RPE65変異に関連するLCA2の治療薬として**ボレチゲン ネパルボベク(voretigene neparvovec-rzyl; 商品名Luxturna)**を承認した。これは眼科領域で初めてのFDA承認遺伝子治療製品である。
組換えアデノ随伴ウイルス(rAAV)ベクターにより正常なRPE65遺伝子のコピーを網膜下に導入する。ランダム化比較第III相臨床試験では有意な視力改善が示された。
ただし、RPE65変異はLCA患者全体の約8%にすぎない。他の変異型に対しては現時点で効果が証明された治療法はない。
現時点で承認されている遺伝子治療(ボレチゲン ネパルボベク / Luxturna)の適応は、両アレル性RPE65変異によるLCA2に限られる。RPE65変異はLCA全体の約8%であり、大多数の患者には適応がない。他の遺伝子型に対する治療は研究段階にある。
LCAの病態生理は、視サイクル(Visual Cycle)の破壊により眼が光情報伝達を行えないことに関連している。
視サイクルは網膜色素上皮(RPE)と神経感覚網膜の間で行われる一連の酵素反応であり、食事由来のビタミンAを代謝して11-シス-レチナール(11-cis retinal)を生成し、光色素を産生する。11-シス-レチナールがなければ光情報伝達カスケードは開始されず、視覚神経信号が視覚野に伝達されない。この一連の反応に関与するタンパク質をコードする遺伝子のいずれかに変異があると、視サイクルが遮断されLCAの症状を引き起こす。
組織病理学的には外網膜・光受容体の関与が示されており、LCAは形成不全ではなく変性プロセスであることが示唆されている。眼内先天的ビタミンA代謝阻害と光受容体変性の関連は依然として不明であり、活発な研究分野である。
LCA関連遺伝子は5つの主要なシグナル伝達ネットワークに分類される2)。
ボレチゲン ネパルボベクの長期追跡評価(NCT00481546、NCT00643747)では、治療後6〜12ヶ月で初期のピークが見られた後、網膜感度・視力・機能的利得を含む臨床的利益が進行性に低下することが報告されている。遺伝子治療の効果持続と改善が今後の課題である。
GUCY2D・AIPL1・CEP290変異に対する遺伝子治療が動物モデルで進行中であり、桿体・錐体光受容体の救済において有望な結果が示されている。これらの研究でも正常な遺伝子を送達するためにウイルスベクターが使用されている。
AIPL1変異マウスモデルにおいて、胚性幹細胞(ES細胞)を網膜下に移植し、光情報伝達機構を再構築・発現させる光受容体置換療法が有望な結果を示している。この研究はまだヒトには拡大されていない。
RPE65変異を持つブリアード犬は、動物モデルで初めて網膜遺伝子治療に成功した例として知られる。rAAVベクターを用いた複数の研究により視力回復が報告され、これらの成果がヒト臨床試験の基盤となった。
LCAの経過は安定(約75%)、進行性悪化(約15%)、改善(約10%)の3パターンに分類される。AIPL1変異は進行性悪化と、RPGRIP1変異は安定した経過と関連する。