ARS 1型
原因遺伝子:PITX2(4q25)
主な異常:前眼部異常、歯牙異常、臍周囲の余剰皮膚・臍帯ヘルニア、頭蓋顔面異常、心血管異常

アクセンフェルト・リーガー症候群(Axenfeld-Rieger syndrome; ARS)は、前眼部形成異常と全身異常を組み合わせた先天性疾患群である。神経堤細胞の遊走・分化の異常が根本病因とされている。
歴史的背景として、1920年にAxenfeldが後部胎生環(Schwalbe線の前方偏位・肥厚)と虹彩突起を記述した。1934〜1935年にRiegerが虹彩低形成・瞳孔偏位・多瞳孔症を追加報告した。現在は以下の3段階で分類されている。
疫学については、有病率は約1/200,000とされてきたが、最近の報告では1/50,000〜100,000との推計もある2)4)。性差はなく、通常は両眼性で乳幼児期に診断されることが多い。
遺伝型分類は以下の通りである。
ARS 1型
原因遺伝子:PITX2(4q25)
主な異常:前眼部異常、歯牙異常、臍周囲の余剰皮膚・臍帯ヘルニア、頭蓋顔面異常、心血管異常
ARS 2型
原因遺伝子:13q14(未確定)
主な異常:前眼部異常、緑内障。全身異常は1型・3型より少ない
ARS 3型
原因遺伝子:FOXC1(6p25)
主な異常:前眼部異常、緑内障、感音性難聴、心房中隔欠損、腎異常、白質病変
FOXC1とPITX2の変異がARSの40〜70%を占める5)。ただしARSの60%では原因遺伝子が未同定であり4)、遺伝的多様性は大きい。
別の大規模解析では、分子診断率は56.5%と報告されている11)。FOXC1変異が20.3%、PITX2変異が17.4%、PAX6変異が10.1%を占め、既知遺伝子で説明できない症例も少なくない11)。
原因遺伝子で区別される。1型はPITX2(4q25)変異で歯牙・臍・顔面骨の異常を伴う。3型はFOXC1(6p25)変異で難聴・心欠損・腎異常・神経異常を伴う。2型は13q14にあるが原因遺伝子は未確定で、前眼部異常と緑内障が主体である。遺伝子検査で確定診断できる。
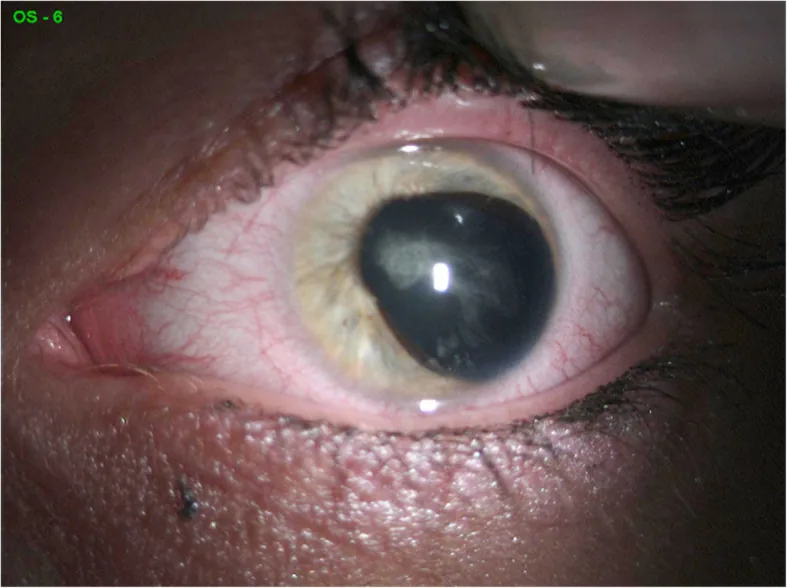
眼所見の主要項目を以下に示す。
| 眼所見 | 特徴 |
|---|---|
| 後部胎生環 | Schwalbe線の前方偏位・肥厚 |
| 虹彩突起 | 細糸状〜幅広帯状 |
| 瞳孔偏位 | 後部胎生環の反対方向へ偏位 |
| 偽多瞳孔 | 虹彩実質の穿孔様外観 |
| ぶどう膜外反 | 虹彩色素上皮の翻転 |
通常、角膜は透明であるが、稀に巨大角膜・小角膜を合併する。FOXC1変異では角膜混濁・角膜血管新生がより顕著であり、PITX2変異と比較して角膜異常の程度が大きく緑内障の頻度も高い1)。小球状水晶体・水晶体亜脱臼を合併する例も報告されている7)。
進行性視力低下を契機に診断される例もあり、前眼部所見と緑内障性変化を見逃さないことが重要である6)。
Li et al.(2021)の7歳男児例(ARS 3型、de novo FOXC1変異)では、角膜径14mm、眼軸長27.16/26.56mm、C/D比0.9、IOP 33/20mmHgを呈した。生後36日から両眼の抗緑内障手術が必要であった5)。
全身所見は以下の通りである。
ARSの約50〜60%に緑内障が合併する。思春期から成人早期にかけて発症することが多いが、乳幼児期から眼圧上昇を来す例もある。定期的な眼圧測定と視神経評価が必要であり、詳しくは「標準的な治療法」の項を参照されたい。
ARSは常染色体優性遺伝を示し、浸透率は完全である。ただし同一家系内で同じ遺伝子変異を持っていても、臨床像に大きな個人差(variable expressivity)が生じる1)。
PITX2遺伝子周辺の微小欠失を有する例では、NEUROG2・UGT8・NDST4の欠失が重なることで発達遅滞・知的障害を合併しうる8)3)。
Kawanami et al.(2023)は、4q25に2.5Mbの微小欠失(PITX2・NEUROG2・ANK2を含む)を有する3歳日本人男児を報告した。臍帯ヘルニア・虹彩コロボーマ・発達遅延を呈したが、ANK2欠失にもかかわらず心電図は正常であった。NEUROG2のハプロ不全が発達遅滞の原因候補とされた8)。
常染色体優性遺伝のため、変異を持つ親からの遺伝確率は50%である。浸透率は完全だが表現型には個人差があり、同じ変異でも症状の重さが大きく異なりうる1)。遺伝子検査と遺伝カウンセリングの受診が推奨される。
ARSの診断は両眼性の隅角異常・虹彩異常に基づく。後部胎生環に周辺虹彩が一部付着していることが診断の条件とされる。細隙灯顕微鏡で後部胎生環が確認できない例では隅角鏡検査が必要である。全身異常を認める場合はARS症候群として小児科への全身精査を依頼する。
なお、正常人口の8〜15%に軽微な後部胎生環が見られるが、これ単独では緑内障等を伴わない。家族歴の聴取も診断において重要である。
ARSと鑑別すべき主な疾患を示す。
| 疾患 | ARSとの相違点 |
|---|---|
| ICE症候群 | 片眼性・後天性・女性優位 |
| Peters異常 | 角膜中央部混濁・Descemet膜欠損 |
| 無虹彩症 | 角膜パンヌス・中心窩低形成 |
| 後部多形性角膜ジストロフィ | 両眼性・家族性・性差なし |
ARSそのものの根治療法は現在存在せず、緑内障の管理と全身合併症のサーベイランスが治療の中心となる。
緑内障はARS症例の約50〜60%に合併する。薬物治療は一般の緑内障に準じて行われる。
房水産生抑制薬
β遮断薬:第一選択の一つ。安全かつ有効。
炭酸脱水酵素阻害薬(点眼):ブリンゾラミドなど。β遮断薬との併用も可。
注意:ブリモニジン(α2作動薬)は2歳未満禁忌。無呼吸・徐脈・低血圧・筋緊張低下・中枢抑制を起こしうる。
房水流出促進薬
プロスタグランジン関連薬:ラタノプロスト、トラボプロストなど。
実例:7歳男児例ではトラボプロスト+ブリンゾラミドで長期管理5)。77歳男性例ではラタノプロスト・チモロール+ブリンゾラミドでもIOP 35mmHgと制御困難であった2)。
プロスタノイドFP受容体作動薬とβ遮断薬の効果に差はないという報告がある。
薬物治療で眼圧制御が得られない場合、手術を行う。治療方針は原発先天緑内障(PCG)に準じる10)13)。
Chakraborty et al.(2022)は、ARS合併網膜剥離(15歳男児)の1例を報告した。小球状水晶体・水晶体亜脱臼を伴い、硝子体手術後にIOP 41mmHgへ上昇し強膜瘤を形成した。ダイオード毛様体光凝固術を施行し最終的にIOP 18mmHgが得られた7)。
隅角手術の成功率はPCGより低い。MMC併用線維柱帯切除術では2年長期成功率は約59%、GDDでは58〜63%とされる。難治例では複数回の手術が必要となることがあり、平均1眼あたり1.5回の手術が報告されている。
ARSの根本的な病因は、神経堤細胞の遊走・分化の欠陥である。前房・前眼部隅角・顔面骨・歯・心血管系・臍周囲皮膚において神経堤細胞の発育が障害されることで多臓器の形成不全が生じる。
組織学的には、角膜後面から前房・隅角・虹彩表面にかけてデスメ膜様の膜を伴う内皮様細胞の単層が異常に広がる。ぶどう膜外反・瞳孔偏位を伴う象限に膜が存在し、反対側の象限では虹彩萎縮が認められる。
FOXC1とPITX2はともに転写因子であり、特定のDNA配列に結合して下流遺伝子の発現を調節する。両者は前眼部発生において相乗的に作用し、共通の下流標的遺伝子を調節する3)。FOXC1のフォークヘッドドメイン(110アミノ酸のDNA結合ドメイン)が機能的に最重要であり2)、このドメインの変異は神経精神症状とより強く関連することが示唆されている。
FOXC1変異マウスでは、角膜実質のコラーゲン線維の減少・構造異常と角膜実質細胞の障害が観察される1)。さらにFOXC1は角膜血管新生の抑制因子として機能し(VEGFの生物学的利用能の制御を介する)1)、FOXC1変異によりこの抑制が失われると角膜血管新生が生じる。
FOXC1はFOXファミリー転写因子として脳の発達にも重要な役割を果たす4)。
系統的レビューでは、ARSの白質異常が41.3%の症例に出現することが報告された4)。FOXC1変異は脳小血管病(CSVD)・白質高信号・拡大血管周囲腔・微小出血・ラクナ梗塞を誘発しうる。
Ohkubo et al.(2025)は、2歳日本人男児(FOXC1変異: c.240del, p.Y81Ifs21)で脳MRIによる脳室周囲白質病変・拡大血管周囲腔・椎骨脳底動脈の迂曲拡張を確認した。父には18歳での脳梗塞の既往があった4)。
FOXC1変異95例のレビューでは6.3%に神経精神症状(学習困難・てんかん・知的障害・嫉妬妄想など)が認められ、フォークヘッドドメイン変異を持つ症例の83.3%に神経精神症状が出現した2)。
はい。FOXC1変異を有するARSでは白質異常が約41%の症例に出現するとの系統的レビューがある4)。FOXC1変異は脳小血管病や脳卒中リスクと関連する可能性が指摘されており、特にFOXC1変異型ARSでは神経学的な長期フォローが重要である。
近年、次世代シーケンシング・全ゲノムシーケンシングにより多くの新規変異が報告されている。
Wowra et al.(2024)は、ポーランド人3姉妹のARSにおいてFOXC1エクソン1の一部と3’UTR全体を含む大規模欠失(新規変異)を同定した。同一家系でも表現型が大きく異なり、当初はChandler症候群と誤診されていた1)。
Jiang et al.(2024)は中国人家族のARS1型においてPITX2を含む6.15Mbの染色体4q25欠失・45.71Mbの逆位・14bpの欠失という複合ゲノム再配列を同定した。11歳女児はIOP 43.5/44.0mmHgを呈した3)。
その他の新規変異の報告として、FOXC1 p.Phe136Leu(フォークヘッドドメイン)2)、FOXC1 p.S82R(de novo変異)5)、FOXC1 c.240del, p.Y81Ifs214)が挙げられる。
Yoshino et al.(2024)は77歳日本人男性のARS3型例を報告した。72歳から嫉妬妄想が出現し、白質脳症を確認した。FOXC1変異95例の文献レビューでは6.3%(6/95例)に神経精神症状が認められ、そのうち83.3%(5/6例)がフォークヘッドドメイン変異を有していた2)。
この知見はFOXC1変異の機能的ドメインが神経精神症状の発現に関与する可能性を示しており、メンタルヘルスの観点からの長期フォローの重要性を示唆している。
FOXC1変異がCSVDを誘導し脳卒中リスクを増加させる可能性が指摘されている4)。ARS患者における神経血管疾患の予防と早期介入の重要性が今後の研究課題とされている。
FOXC1変異による角膜「強膜化」の病態解明が進んでいる1)。角膜混濁の分子メカニズムの理解が、遺伝子治療・抗線維化薬・生体材料を用いた新規治療開発へ繋がりうると期待されている1)。