初期
RPE萎縮と色素沈着:赤道部・中間周辺部から始まる顆粒状のRPE萎縮。周辺部に色素沈着を伴う。
脈絡膜毛細血管板の消失:周辺部から始まり、中間周辺部へ向かって進行する。

コロイデレミア(Choroideremia)は、CHM遺伝子の変異によって生じるX連鎖劣性遺伝性の網脈絡膜ジストロフィである。網膜色素上皮(RPE)・視細胞・脈絡膜毛細血管板の三層が広範かつ進行性に変性する。
1872年にLudwig Mauthnerが初めて報告した。病名は古代ギリシャ語の「chorion(皮)」と「eremia(不毛の地)」に由来する。
有病率は5万〜10万人に1人と推定される3)。主に男性が罹患し、女性は通常無症状の保因者となる。末期には脈絡膜がほぼ完全に消失し、白い強膜が眼底全体に露出する。網膜色素変性の類縁疾患として分類される。
CHM遺伝子はXq21.2に位置し、186,382 bpの領域にわたる。15エキソン、ORF 1,962 bpからなり、653アミノ酸・95 kDaのRab escort protein 1(REP-1)をコードする。病原性変異は280以上が報告されており5)、その多様性が高い。最も頻度の高い5変異を合計しても全変異の11%にすぎない1)。
X連鎖劣性遺伝のため、発症は主に男性である。女性保因者は通常無症状だが、ランダムなX染色体不活化のパターンによっては高齢になると視機能異常が現れる場合がある。保因者には斑点状のモザイク状眼底変化が観察されることがある6)。
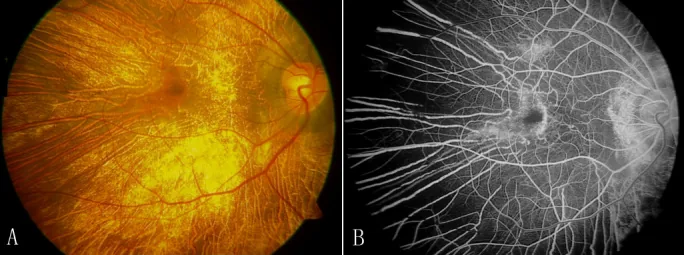
病期によって特徴的な眼底所見を呈する。
初期
RPE萎縮と色素沈着:赤道部・中間周辺部から始まる顆粒状のRPE萎縮。周辺部に色素沈着を伴う。
脈絡膜毛細血管板の消失:周辺部から始まり、中間周辺部へ向かって進行する。
中期
島状脈絡膜萎縮:斑紋状・島状の萎縮巣が融合する。脈絡膜大血管が透見される。
残存脈絡膜の求心性萎縮:萎縮縁が中心方向へ進行する。
末期
びまん性脈絡膜萎縮:脈絡膜がほぼ完全に消失し、白い強膜が露出する(白色眼底)。
foveal sparing:中心窩の組織が島状に最後まで残存する1)7)。
画像検査では以下の所見が特徴的である。
主な画像検査所見を以下に示す。
| 検査 | 特徴的所見 |
|---|---|
| FAF | 周辺部の自発蛍光消失が求心性に進行。消失率は年7.7%2) |
| OCT | 脈絡膜菲薄化、RPE消失、エリプソイドゾーン消失、外顆粒層の網膜細管構造2) |
| 網膜電図 | 初期から全反応で著明な減弱または消失8) |
蛍光眼底造影(FA)では脈絡膜毛細血管板消失部位が低蛍光を呈し、残存灌流部位が過蛍光を示す。OCT血管造影(OCTA)では脈絡膜血管密度の低下が視細胞喪失に先行する所見が得られる2)。
女性保因者では斑点状脱色素斑がモザイク状に散在する6)。保因者に卵黄様病変が生じることも報告されている6)。稀な合併症として網膜分離症5)および脈絡膜新生血管(CNV)4)がある。
Greigら(2022)は、コロイデレミア患者における網膜分離症が網膜剥離に類似した外観を呈し、マルチモーダル画像検査により鑑別可能であることを報告した5)。OCT・OCTAを用いることで、正確な診断と不必要な手術回避に貢献する。
コロイデレミアの原因はCHM遺伝子の機能喪失型変異である。変異の約30%がナンセンス変異で占められる2)。CHM変異のほぼすべてが機能喪失型であり1)5)、残存REP-1機能はほぼゼロとなる。
X連鎖劣性遺伝のため、主なリスク因子は男性であることと家族歴(母方の保因者)である。
大規模なXq21領域欠失(約13.5 Mbp・18遺伝子以上に及ぶ欠失)では、コロイデレミアに加え発達遅滞(78%)や難聴(56%)を伴う症候性コロイデレミアを呈する3)。
280以上の病原性変異が報告されており、家族固有の変異が多い。最頻5変異を合わせても全病原性変異の11%にすぎず1)、変異の多様性が高い。約30%がナンセンス変異である2)。アタルレンなどナンセンス変異特異的治療の対象となりうる患者群の同定に、変異の種類の把握が重要である。
特徴的な眼底所見と家族歴が診断の手がかりとなる。確定診断には以下のいずれかを用いる。
OCT検査では、網膜分離症と網膜剥離の鑑別が重要な場面がある5)。マルチモーダル画像検査(FA・FAF・OCT・OCTA)の組み合わせが病態評価と病期判定に有用である。
主な鑑別疾患を以下に示す。
| 疾患 | 遺伝形式 | 鑑別のポイント |
|---|---|---|
| 脳回状脈絡膜萎縮 | AR | 高オルニチン血症 |
| 網膜色素変性 | 多様 | 骨小体様色素沈着 |
| びまん性脈絡膜ジストロフィ | AD | 40〜50代発症 |
コロイデレミアでは境界の鮮明な脈絡膜萎縮巣と末期の白色眼底が特徴的である。網膜色素変性では骨小体様色素沈着と赤道部への色素沈着パターンが異なる。X連鎖劣性遺伝形式と家族歴を確認したうえで、CHM遺伝子検査によって確定診断する。
現時点では、コロイデレミアに対して承認された有効な治療法は存在しない。治療は合併症管理と定期的な経過観察が中心となる。
遺伝子治療・細胞治療などの研究が進んでいる。詳細は「最新の研究と今後の展望」の項を参照。
2025年時点で承認された遺伝子治療薬はない。Phase III試験(STAR trial)でprimary endpoint(3ライン改善)は未達であったが1)、2ライン改善では有意差が認められた1)。現在もエンドポイントの再設定や新規試験が進行中であり、今後の承認に期待がかかる。
コロイデレミアの病態はREP-1(Rab escort protein 1)の欠損に起因する小胞輸送障害である。
REP-1はRab GTPaseのプレニル化(ゲラニルゲラニル基の付加)を促進するシャペロンタンパク質である1)。プレニル化によりRabタンパク質に疎水性が付与され、細胞内膜への結合が可能となる1)。Rabタンパク質はエンドソーム・リソソームの小胞輸送に必須であり、RPE細胞における視細胞外節のファゴサイトーシスを含む多くの細胞機能を担う1)。
CHM変異によりREP-1が欠損すると、Rabタンパク質のプレニル化が障害される1)。部分的な代償としてREP-2が機能するが、Rab27aなど一部のRabタンパク質はREP-1を優先的に利用するため、代償は不完全である2)。
小胞輸送障害の結果、RPE細胞内にリポフスチンが蓄積し1)、メラノソームが減少する1)。「まず細胞機能障害、後に細胞死」というパターンを経てRPE細胞が脱落し、二次的に視細胞・脈絡膜毛細血管板の変性が進行する1)。
変性が赤道部・中間周辺部から始まる機序として、その部位でのRPE細胞あたりの視細胞密度が最大であることが指摘されている1)。代謝負荷が最も高い部位から先に障害が生じると考えられる。中心窩では視細胞密度が相対的に低く、この部位が最後まで保存される(foveal sparing)1)7)。
コロイデレミアは「加齢黄斑変性など加齢性眼疾患の原型」とも称される1)。正常老化でも生じるRPEの機能低下と類似した機序が、若年期から進行することが本疾患の特徴である。
網膜分離症の合併については、REP-1欠損によるRPEの分泌機能障害が関与すると推定されている5)。
コロイデレミアの遺伝子治療は最も進展した分野である。CHM cDNA(1.96 kb)のサイズがAAVの積載能力に適合しており1)、AAV2ベクターによる網膜下投与が標準的手技となっている。手術は、BSSを網膜下腔に注入して局所網膜剥離を誘発し、その後AAV溶液を注入する方法で行われる1)。
複数のPhase I/II試験が実施されており、40患者の全体中央値ETDRS変化は+1.5であった2)。Oxfordグループの14患者では中央値+5.5のETDRS改善が報告されている2)。
Phase III試験(timrepigene emparvovec / STAR trial)では140名が登録された。Primary endpoint(3ライン改善)は未達であったが、高用量群での視力変化は-0.3 ETDRS(対照群-2.3 ETDRS)であり、2ライン改善では有意な効果が認められた1)。
REGENERATE試験では、早期段階の患者においてsmooth zone(SW-AFでの均一自発蛍光領域)の保存が観察されている1)。今後の試験ではprimary endpointとして2ライン改善を採用する提案がなされている1)。
代替遺伝子治療
4D-110(硝子体内投与型):網膜下注射を要しない硝子体内投与ベクター。NCT04483440で試験中2)。
アタルレン(PTC124):ナンセンス変異を持つ約30%の患者を対象に、リードスルー効果を狙った治療2)。
細胞・ゲノム編集
iPSC由来RPE移植:幹細胞から分化させたRPEの移植研究が進行中2)。
CRISPR:CHM cDNAの遺伝子置換は原理実証段階。手術ロボットとの併用も研究中1)。
症候性コロイデレミア(Xq21大規模欠失)に関しては、欠失範囲と表現型の関係の詳細な解析が進み、遺伝学的理解が深まっている3)。