眼科的所見

ヌーナン症候群
ひとめでわかるポイント
Section titled “ひとめでわかるポイント”1. ヌーナン症候群とは
Section titled “1. ヌーナン症候群とは”ヌーナン症候群(Noonan syndrome; NS)は、RAS-MAPK(マス・マップキナーゼ)シグナル経路の遺伝子変異を原因とする遺伝性疾患(RASopathy)である。1968年にJacqueline Noonanにより初めて体系的に報告された。
推定有病率は出生1,000〜2,500人に1人とされる。遺伝形式は通常、常染色体顕性遺伝(AD)であるが、症例の約2/3は新規(de novo)変異による。ADおよびAR(常染色体潜性遺伝)の両形式をとるLZTR1変異が知られており、Zhao et al.の中国家系ではLZTR1 c.1149+1G>T変異がAD遺伝を示した1)。
同一変異を持つ家系内でも表現型が大きく異なる「可変表現度」と、保因者が表現型を示さない「不完全浸透」が特徴的である。Han & Park(2024)はPTPN11 p.Arg498Trp変異を持つ父系遺伝例を報告しており、父親は無症状〜軽度知的障害のみで、罹患親族は30〜40%とされる2)。de novo変異は父親の高年齢と関連し、精子形成過程で生じた変異が選択的優位性を持つと考えられている。
眼科的には、視神経低形成・視神経乳頭コロボーマ・屈折異常・眼瞼下垂など多彩な異常を伴いうる。
Q ヌーナン症候群はどのくらい遺伝するのか?
A
通常は常染色体顕性遺伝を示すが、約2/3はde novo変異(自然発生)による。罹患した親から子への遺伝は理論的に50%の確率であるが、不完全浸透のため実際に症状が現れる割合は30〜40%とされる2)。家族歴がある場合は遺伝カウンセリングが推奨される。
2. 主な症状と臨床所見
Section titled “2. 主な症状と臨床所見”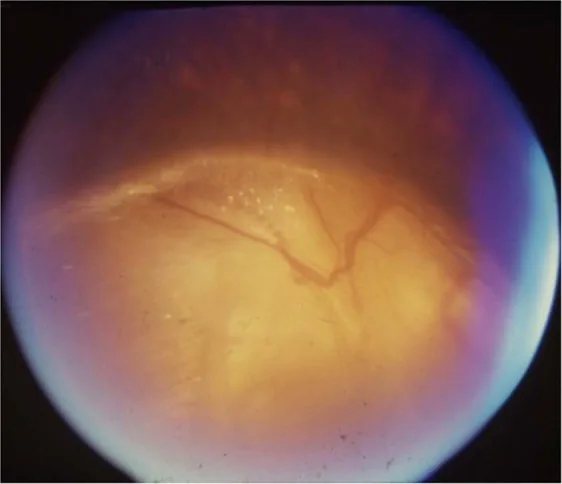
Gopal Lingam, Alok C Sen, Vijaya Lingam et al. Ocular coloboma—a comprehensive review for the clinician. Eye. 2021 Mar 21; 35(8):2086. Figure 3. PMCID: PMC8302742. License: CC BY.
?????????????????????????????????????????Noonan???????????????????????
- 視力低下:視神経低形成やコロボーマに伴い、乳児期から視力不良の場合がある。
- 斜視:眼位のずれを自覚する。小児期に親が気づくことが多い。
- 低身長:出生時身長は正常だが、成長とともに低身長が明らかとなる。
- 発達遅滞:言語発達の遅れ・軽度知的障害が初期の主訴となることがある。
全身所見
特徴的顔貌:広い額、鼻根部陥凹、肉厚な耳輪を伴う低位後方回旋耳。乳児期・小児期に最も顕著。
心血管系:肺動脈弁狭窄症(50〜60%)、肥大型心筋症(20%)、ASD(6〜10%)、VSD、リンパ管形成不全。
骨格・頸部:翼状頸、鳩胸・漏斗胸、乳頭間離解、相対的大頭症。
血液学的:PT/PTT延長、血小板数・機能異常、第XI因子欠乏(50%)。新生児期の難治性血小板減少が初発症状となりうる5)。
生殖器:男性の停留精巣(最大80%)、不妊症。
Zhao et al.の症例(6.6歳女児)では斜視・屈折異常・眼振が認められ、成長ホルモン欠乏を合併していた1)。Tian et al.(2025)はLZTR1変異NSにおいて門脈海綿状変化・腸リンパ管拡張・蛋白漏出性腸症を報告しており、リンパ管形成異常の多彩な病態が示された6)。
Q ヌーナン症候群の眼の症状にはどのようなものがあるか?
A
屈折異常(近視・遠視・乱視)、眼瞼下垂、眼位のずれ(斜視)、眼振、弱視など多彩な所見を呈する。より重篤な所見として、視神経低形成・視神経乳頭コロボーマ・円錐角膜が認められる場合がある。これらは診断時から包括的眼科評価で評価することが推奨される。
3. 原因とリスク要因
Section titled “3. 原因とリスク要因”NSの原因は、RAS-MAPK経路に関与する複数の遺伝子の機能獲得型(または機能喪失型)変異である。原因遺伝子の頻度を以下に示す。
| 遺伝子 | 頻度 | 主な特徴 |
|---|---|---|
| PTPN11 | 約50% | 最多。機能獲得型変異が主 |
| SOS1 | 約10〜13% | — |
| RAF1, RIT1 | 各約5% | 視力障害との関連報告あり |
| LZTR1 | 約8% | AD・AR両遺伝形式 |
| KRAS, BRAF | 各5%未満 | BRAF変異で最低視力の報告 |
LZTR1はBTB-Kelchスーパーファミリーに属するゴルジ体蛋白であり、RASのユビキチン化・分解を促進してRAS-MAPKシグナルを負に制御する3)。AD-NSの変異はKelchドメイン(基質認識面)に集中し、RAS-MAPKシグナルを亢進させる1)3)。
遺伝子型と眼科的表現型の相関については、RAF1・KRAS・SHOC2変異患者で恒久的視力障害(両眼最高矯正視力 < 0.3)、BRAF変異患者で最低視力(視神経低形成による乳児期からの視力不良)の報告があるが、確定的な相関は未確立でありコホートが小規模(n=25〜105)である。
NF1変異によるNFNS(神経線維腫症-ヌーナン症候群)では、NF1患者の約25%がNoonan様特徴を示し、両疾患の表現型が重複する4)。
Q 遺伝子の種類によって目の症状の重さは変わるのか?
A
RAF1・KRAS・SHOC2変異患者では恒久的な視力障害の報告があり、BRAF変異患者では視神経低形成による乳児期からの視力不良が報告されている。一方、PTPN11変異患者では視力障害は認められなかったとする報告もある。ただし、これらの相関を示すコホートは小規模であり、現時点で確定的な遺伝子型-表現型相関は確立されていない。
4. 診断と検査方法
Section titled “4. 診断と検査方法”臨床診断・遺伝学的検査
Section titled “臨床診断・遺伝学的検査”NSの診断は、特徴的顔貌・心疾患・低身長などの臨床所見に基づく正式な診断基準に従い、臨床遺伝専門医が評価する。
確定診断にはRAS-MAPK経路関連遺伝子パネル(PTPN11, SOS1, RAF1, RIT1, KRAS, LZTR1等)による分子遺伝学的検査を用いる2)3)。全エクソームシーケンス(WES)も使用される1)4)。NSの遺伝子パネルにLZTR1が含まれていない場合があるため注意が必要である3)。
診断時に包括的眼科評価を行い、その後は年1回のフォローアップが推奨される。
- 屈折検査:屈折異常の評価。強度屈折異常・乱視の有無を確認。
- 斜視検査:眼位および眼球運動の評価。
- 眼底検査:視神経乳頭の形態評価。
視神経低形成の評価
Section titled “視神経低形成の評価”- DM/DD比(乳頭黄斑間距離/乳頭径比):3以上で視神経低形成を疑い、4以上で可能性が高い。
- double ring sign:乳頭周囲の色素輪。
- 光干渉断層計(OCT):cpRNFL(乳頭周囲網膜神経線維層)厚測定が有用。
- 頭部MRI:中枢神経系異常の評価。約15%に下垂体漏斗異常を認める。
視神経乳頭コロボーマの診断
Section titled “視神経乳頭コロボーマの診断”- 検眼鏡所見:下方を中心とした乳頭・網脈絡膜の欠損、血管走行異常。
- 画像検査:エコー・MRI・CT・OCTによる確定診断。頭部MRI/CTで頭蓋内奇形合併の有無を検索。
- NFNS(神経線維腫症-ヌーナン症候群):NF1変異によりNS+NF1の表現型が重複。cafe-au-lait斑6個以上またはサイズ5mm以上でNF1合併を検索4)。Farncombe et al.はLZTR1変異NS患者に叢状神経線維腫を認め、NF1との臨床的オーバーラップを報告した3)。
- 視神経乳頭コロボーマの鑑別:乳頭周囲ぶどう腫、朝顔症候群、乳頭部PFV/PHPV、巨大乳頭症。
- その他の症候群:CHARGE症候群、Turner症候群、心顔面皮膚症候群(CFC)、Costello症候群、Legius症候群。
その他の検査
Section titled “その他の検査”- 心エコー:心奇形スクリーニング。
- 血液検査:凝固機能検査(PT/APTT、第XI因子活性など)5)。
5. 標準的な治療法
Section titled “5. 標準的な治療法”NSは根治療法がなく、臨床遺伝専門医を中心とした多職種連携による集学的管理が基本となる。
- 屈折矯正:屈折異常に対する眼鏡・コンタクトレンズによる矯正。
- 弱視治療:健眼遮蔽などを用いた弱視の治療。
- 斜視手術:斜視の程度に応じて手術を検討する。
- 視神経低形成:非進行性であり、緑内障を合併しなければ視力・視野は変化しない。安易な眼圧下降治療は慎む。適切な屈折矯正で残存視機能の改善を試みる。
- 視神経乳頭コロボーマに伴う漿液性網膜剥離:定まった治療法はなく、自然消退例もある。裂孔原性網膜剥離にはそれに準じた手術療法を検討する。
- 低身長:成長ホルモン(GH)療法。NS患者でGH欠乏が合併する例あり1)。ただしNFNS1ではGH投与で神経線維腫の増大リスクがあり慎重な判断が必要4)。
- 心疾患:肺動脈弁狭窄症に対するバルーン拡張術・外科的修復。肥大型心筋症の経過観察・薬物治療。2歳未満の肥大型心筋症は心臓死の最高リスクとなる5)。
- 血液異常:凝固因子補充。手術前の出血リスク評価。Tang et al.の新生児例では難治性血小板減少が初発症状であり、ASDと乳糜胸を合併していた5)。
- 乳糜胸:胸腔ドレナージ。Tian et al.では胸管出口閉塞に対する顕微鏡下手術で低蛋白血症の改善が得られた6)。
- 停留精巣:適切な時期の精巣固定術。
Q ヌーナン症候群の目の治療はどのように行われるか?
A
眼鏡・コンタクトレンズによる屈折矯正と弱視治療が基本となる。斜視は程度に応じて手術を検討する。視神経低形成は非進行性であり、安易な眼圧下降治療は慎み、適切な屈折矯正で残存視機能の改善を図る。視神経乳頭コロボーマに伴う漿液性網膜剥離は自然消退例もあり、定まった治療法はない。
6. 病態生理学・詳細な発症機序
Section titled “6. 病態生理学・詳細な発症機序”NSの根本的病態生理は、RAS-MAPK経路の調節不全による持続的・過剰なシグナル伝達である。
PTPN11(SHP2)
機能:非受容体型チロシンホスファターゼ。N-SH2ドメインとPTPドメインの分子内相互作用による自己抑制機構を持つ。
NS変異の影響:自己抑制機構が解除され、ホスファターゼ活性が亢進(機能獲得型)。RAS-MAPKシグナルが過活性化する2)。
NSMLとの相違:NSML( leopard症候群)変異はPTPN11の活性低下型であり、NSとは対照的である。
LZTR1
機能:BTB-Kelchスーパーファミリーに属するゴルジ体蛋白。RASのポリユビキチン化・プロテアソーム分解を促進し、RAS-MAPKシグナルを負に制御する3)。
AD-NSの変異:Kelchドメイン(基質認識面)に集中。RAS-MAPKシグナルを亢進させる(機能獲得型)1)3)。
AR-NSの変異:機能喪失型変異による。RAF1/SHOC2/PP1CB複合体に結合しRAF1 Ser259リン酸化を促進する(MAPKシグナル不活化)機能も有する3)。
RAS-MAPK経路の主要構成要素は以下の通りである。
- 正の調節因子:SHP2(PTPN11)、SOS1、SOS2。これらの機能獲得型変異がNSの原因となる。
- RASシグナル促進因子:MRAS、SHOC2、PPP1CB。
- MAPKカスケード:BRAF、RAF1、MAP2K1、MAP2K2、MAPK1。
- 負の調節因子:CBL、NF1、LZTR1、SPRED1、SPRED2。機能喪失型変異がシグナル亢進の原因となる3)。
RAS-MAPK経路は細胞の分化・増殖に極めて重要であり、機能獲得型変異により全身組織で異常な細胞機能が生じ、多臓器にわたる複雑な表現型をもたらす。
眼科的病態としては、視神経低形成は網膜神経節細胞・神経線維の発生不全による。視神経乳頭コロボーマは眼杯裂の閉鎖不全に起因し、有病率は3〜8/100,000とされる。
腫瘍との関連として、PTPN11変異はJMML(若年性骨髄単球性白血病)・MDS・AMLの体細胞変異としても同定されており、NSは小児期の血液悪性腫瘍リスクが3倍とされる5)。
7. 最新の研究と今後の展望(研究段階の報告)
Section titled “7. 最新の研究と今後の展望(研究段階の報告)”LZTR1変異NSにおけるリンパ管形成異常
Section titled “LZTR1変異NSにおけるリンパ管形成異常”Tian et al.(2025)はLZTR1 c.850C>T変異NSの症例において、門脈海綿状変化・胸管形成不全・腸リンパ管拡張・蛋白漏出性腸症を報告した6)。胸管出口閉塞に対する顕微鏡下手術を施行し、血清アルブミンの正常化を達成した。これはLZTR1変異NSに伴うリンパ管形成異常の新たな病態を示す知見である。
男性不妊の機序
Section titled “男性不妊の機序”Orsolini et al.(2024)はLZTR1 c.742G>A変異を持つ35歳男性において、停留精巣の既往なく高FSH・乏精子症(精子濃度1.5×10⁶/mL)を認めたことを報告した7)。この症例は停留精巣とは独立した原発性Sertoli細胞障害による性腺機能障害の存在を示唆しており、NSにおける男性不妊の機序解明に寄与する知見である。
遺伝子型-表現型相関と今後の課題
Section titled “遺伝子型-表現型相関と今後の課題”LZTR1は神経鞘腫症(schwannomatosis)の原因遺伝子でもあり、NS・NF1・神経鞘腫症の臨床的オーバーラップに関する研究が進んでいる3)。RIT1とLZTR1の生物学的関係についても注目されており、どちらかの変異がRIT1蓄積を引き起こしMAPKシグナルの過活性化に寄与する可能性が示唆されている3)。
遺伝子型-表現型相関については、より大規模なコホートによる確立が今後の課題である。眼科的予後の予測因子の同定や、NS特異的な治療標的の開発が期待される。
8. 参考文献
Section titled “8. 参考文献”- Zhao X, Li Z, Wang L, Lan Z, Lin F, Zhang W, Su Z. A Chinese family with Noonan syndrome caused by a heterozygous variant in LZTR1: a case report and literature review. BMC Endocr Disord. 2021;21(1):2.
- Han JY, Park J. Paternally Inherited Noonan Syndrome Caused by a PTPN11 Variant May Exhibit Mild Symptoms: A Case Report and Literature Review. Genes. 2024;15(4):445.
- Farncombe KM, Thain E, Barnett-Tapia C, Sadeghian H, Kim RH. LZTR1 molecular genetic overlap with clinical implications for Noonan syndrome and schwannomatosis. BMC Med Genomics. 2022;15(1):160.
- Dalili S, Hoseini Nouri SA, Bayat R, Koohmanaee S, Tabrizi M, Zarkesh M, Tarang A, Mahdieh N. Neurofibromatosis-Noonan syndrome and growth deficiency in an Iranian girl due to a pathogenic variant in NF1 gene. Hum Genomics. 2023;17(1):12.
- Tang X, Chen Z, Shen X, Xie T, Wang X, Liu T, Ma X. Refractory thrombocytopenia could be a rare initial presentation of Noonan syndrome in newborn infants: a case report and literature review. BMC Pediatr. 2022;22(1):142.
- Tian QJ, Zhang LJ, Zhang Q, Liu FC, Xie M, Cai JZ, Rao W. Protein-losing enteropathy and multiple vasculature dysplasia in LZTR1-related Noonan syndrome: A case report and review of literature. World J Gastroenterol. 2025;31(17):105347.
- Orsolini F, Pignata L, Baldinotti F, Romano S, Tonacchera M, Canale D. Gonadal dysfunction in a man with Noonan syndrome from the LZTR1 variant: case report and review of literature. Front Endocrinol. 2024;15:1354699.