Type I
角膜後面欠損と角膜混濁:Descemet膜・角膜内皮が限局性に欠損する。角膜虹彩癒着を伴う。水晶体は正常位に存在する。最も多い病型である1)。
Peters異常(Peters anomaly)は、角膜内皮、Descemet膜、角膜実質の一部が先天的に欠損し、角膜中央部に円板状の混濁をきたす先天異常である。胎齢6週以降の神経堤細胞の遊走異常に起因する。約80%は両眼性であり、緑内障を50〜70%に合併する。
発生頻度は約1.5/100,000出生と報告されている1)。先天性角膜混濁(congenital corneal opacity: CCO)の最多原因であり、CCO全体の40.3〜65%を占める3)。角膜後面欠損と角膜混濁のみのType I、角膜水晶体癒着を伴うType II、および全身異常を伴うPeters Plus症候群に分類される1)。
Khasnavisらの総説では、Peters異常はCCOの中で最も高頻度の原因であり、重症度に基づくステージ分類(Stage 1〜5)が提唱されている1)。Stage 1は中央3 mm未満の混濁、Stage 5は角膜全体の混濁と虹彩癒着を伴う最重症型である。
重症例では角膜全体が前方へ突出する前部ぶどう腫を呈する。低身長、骨格異常、口唇・口蓋裂などの全身異常を約1/3の症例に伴う。全身的な先天奇形の合併として、中枢神経系異常、口蓋・口唇奇形、心奇形、肺低形成、泌尿生殖器異常、脊髄披裂、仙骨低形成、13トリソミーや15トリソミーなどがあり、Peters異常を診察したときには全身検索を行う。
European Glaucoma Society(EGS)のガイドラインでは、Peters異常は非後天性眼異常に伴う小児緑内障の代表的原因として記載されている7)。また、AAOのCorneal Edema and Opacification Preferred Practice Patternでも、角膜混濁の先天性原因としてPeters異常が列挙されている8)。
大部分の症例は孤発性(散発性)である2)。ただし、PAX6、PITX2、FOXC1、FOXE3、CYP1B1などの遺伝子変異が関与する家族性の症例も報告されている。Peters Plus症候群はB3GLCT遺伝子の常染色体劣性変異による。遺伝カウンセリングの際には、表現型の多様性と浸透率の不完全性を考慮する必要がある。
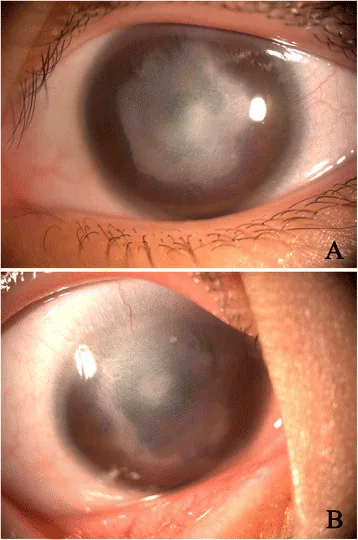
角膜中央部の円板状混濁が最も特徴的な所見である。Type IとType IIで所見が異なる。
| 所見 | 特徴 |
|---|---|
| 角膜中央部混濁 | Descemet膜と内皮の欠損部位に一致 |
| 角膜虹彩癒着 | Type Iで認める |
| 角膜水晶体癒着 | Type IIで認める |
Khasnavisらは重症度に基づく5段階のステージ分類を示している1)。Stage 1(中央3 mm未満の混濁)から、Stage 5(角膜全体の混濁+虹彩癒着)まで段階的に重症度が増す。組織学的には、Descemet膜欠損部に線維性組織が認められる1)。
緑内障を合併した場合は角膜浮腫の増悪、牛眼(眼球拡大)を呈する。重症例では前部ぶどう腫を認める。
Type I
角膜後面欠損と角膜混濁:Descemet膜・角膜内皮が限局性に欠損する。角膜虹彩癒着を伴う。水晶体は正常位に存在する。最も多い病型である1)。
Type II
角膜水晶体癒着:角膜混濁に加え、水晶体が角膜後面に癒着する。白内障を高率に合併する。Type Iより重症であり、視力予後も不良である1)。
Peters Plus症候群
全身異常の合併:Peters異常に低身長、口唇・口蓋裂、短指症を伴う症候群である。B3GLCT遺伝子の常染色体劣性変異が原因である2)。
Peters異常に関連する遺伝子は多数同定されている。前眼部形成異常(anterior segment dysgenesis: ASD)全体の遺伝子解析において、FOXC1変異が最多(20.3%)、次いでPITX2(17.4%)、PAX6(10.1%)の順に多い6)。PITX2変異を有する症例の大部分はAxenfeld-Rieger症候群を呈するが、1例でPeters異常が報告されている6)。
| 遺伝子 | 関連する表現型 |
|---|---|
| PAX6 | PA、無虹彩 |
| FOXC1 | PA、ARS |
| PITX2 | PA、ARS |
| FOXE3 | PA、白内障 |
| CYP1B1 | PA、先天緑内障 |
Cronbachらは、非症候群性小児緑内障およびASDに関連する20の原因遺伝子を報告した。Peters異常に関連する遺伝子としてCYP1B1、FOXC1、PAX6、PITX2、FOXE3に加え、COL4A1、PXDN、SOX11、CDH2、KDM5C、TFAP2Aが同定されている4)。
Kaushikらは、Peters異常がDescemet膜と角膜内皮の欠損を特徴とし、罹患者の約半数が緑内障を発症すると報告している5)。
Peters Plus症候群は、Peters異常に加えて低身長、口唇・口蓋裂、短指症(中手骨・中足骨の短縮)などの全身異常を合併する症候群である2)。B3GLCT遺伝子の常染色体劣性変異が原因で、糖転移酵素の機能障害により多臓器の発生異常をきたす。EGSガイドラインでも全身異常を伴う場合はPeters Plus Syndromeとして区別している7)。
| 疾患 | 鑑別のポイント |
|---|---|
| 先天緑内障 | 牛眼、Haab線条を伴う |
| CHED | 両眼性角膜浮腫。内皮異常 |
| 強膜化角膜 | 輪部が不明瞭。角膜全体白濁 |
先天緑内障との鑑別が特に重要である。先天巨大角膜では眼圧上昇やHaab線条を認めず、隅角は正常である。Axenfeld-Rieger症候群との鑑別が困難な症例もあり、遺伝子検査が診断の補助となる5)。
角膜混濁は緩徐に軽減することが多く、眼圧に注意しながら経過観察を行う。高眼圧を認めた場合、あるいは角膜径が次第に増大する場合には、まず抗緑内障点眼薬を処方し、無効な場合に線維柱帯切開術などの観血的治療を行う。角膜移植術後の経過不良もあるため、本疾患に対しては移植をせず経過観察を行うことが多い。角膜混濁が軽減して不正乱視が高度な場合にはハードコンタクトレンズによる矯正を試みる。重症例で視覚発達への影響が大きい場合に外科的介入を検討する。
全層角膜移植術(PKP)
適応:重症の角膜混濁で視力発達が阻害される場合。
成績:移植片透明率は39〜90%と報告に幅がある1)。拒絶反応が移植片不全の主因であり、不全例の65%を占める3)。小児では拒絶反応の可逆率が27〜28%と成人より低い3)。
課題:術後の弱視管理が不可欠である。
選択的角膜内皮除去術(SEPA)
概要:混濁部のDescemet膜と角膜内皮を選択的に除去し、周囲の健常内皮細胞による再被覆を期待する低侵襲手技である1)。
成績:34眼中85%で角膜透明化が得られたと報告されている1)。
利点:ドナー角膜が不要であり、拒絶反応のリスクがない。
その他の外科的選択肢
光学的虹彩部分切除術:混濁の周辺にある透明部分を通して光路を確保する方法である3)。
角膜内皮移植術(DSAEK):角膜内皮機能障害が主体の場合に考慮される層状移植術である3)。
角膜プロテーゼ:Boston type 1 KProが複数回の移植片不全例に対して用いられることがある3)。
緑内障は20〜50%に合併し1)、視力予後を左右する最重要合併症である。薬物治療を第一選択とし、コントロール不良であれば流出路再建術(線維柱帯切開術)や濾過手術(線維柱帯切除術)を行う。角膜混濁のため眼圧測定が困難な場合は超音波生体顕微鏡による隅角評価が術式選択の参考となる。
EGSガイドラインでは、治療は原疾患と眼圧上昇のメカニズムに合わせて個別化し、可能な限り三次医療施設への紹介を推奨している7)。
SEPA(selective endothelialectomy with peripheral autokeratoplasty)は、混濁部位のDescemet膜と角膜内皮のみを選択的に除去する手技である1)。ドナー角膜を使用せず、患者自身の周辺部の健常な角膜内皮細胞が欠損部を再被覆することで角膜の透明化を図る。34眼中85%で角膜透明化が報告されており1)、拒絶反応のリスクがなく、ドナー不足の問題も回避できる利点がある。ただし、周辺部に十分な健常内皮が残存している症例が対象となる。
視覚発達の臨界期は生後2〜6か月であり、重症の両眼性角膜混濁では早期介入が望ましい。しかし、眼圧が正常な軽症例では角膜混濁が自然に改善する可能性があるため、経過観察が選択されることが多い。手術のリスクと弱視予防の利益を個別に評価し、角膜専門医と小児眼科医の連携のもとで時期を決定する。
Peters異常は、胎齢5〜7週目に起こる神経堤細胞(neural crest cell: NCC)の遊走異常に起因する4)。NCCは3つの波で前眼部に遊走し、それぞれ角膜内皮、虹彩実質、角膜実質の形成に寄与する。
第1波のNCCは角膜内皮とDescemet膜を形成する。この波の遊走障害がPeters異常の中核的病態である。Descemet膜と角膜内皮の欠損により、角膜実質の透明性が維持できず、中央部に円板状の混濁を生じる1)。
第2波のNCCは虹彩実質と瞳孔括約筋を形成する。この波にも異常が及ぶと、虹彩が角膜後面に癒着する(Type I)。さらに水晶体の分離にも異常が生じると、水晶体が角膜後面に癒着する(Type II)4)。
PAX6は眼杯形成に不可欠な転写因子である4)。FOXC1とPITX2は前眼部間充織(periocular mesenchyme)の分化を制御する2)。これらの遺伝子の変異はNCCの遊走・分化を障害し、Peters異常を含む前眼部形成異常スペクトラムを引き起こす。
Paredesらは、前眼部形成異常の遺伝学的アプローチとして、表現型(角膜混濁のパターン、虹彩癒着の有無)から候補遺伝子を絞り込む診断アルゴリズムを提唱した2)。Peters異常ではPAX6、FOXE3、PITX2、FOXC1、PITX3が主要な候補遺伝子である。
CYP1B1はチトクロームP450ファミリーに属し、原発先天緑内障(PCG)の主要原因遺伝子であるが、Peters異常にも関与する4)5)。FOXE3の変異は水晶体分離の異常を引き起こし、Peters異常Type IIの病態に関連する2)。
SEPAは、ドナー角膜を使用せず角膜内皮の選択的除去のみで角膜透明化を図る画期的な手技である1)。34眼中85%で透明化が得られた報告は有望であるが、長期成績と適応基準の確立が今後の課題である。
次世代シーケンシング(NGS)技術の発展により、ASD関連遺伝子の包括的解析が可能となっている。Childhood Glaucoma Research Network(CGRN)による大規模遺伝子解析では、ASD全体の遺伝子診断率の向上が報告されている6)。Paredesらの表現型→遺伝子型アルゴリズムは、臨床現場での効率的な遺伝子検査の実施に貢献しうる2)。
CRISPRを用いた遺伝子治療はMYOC変異による緑内障で研究が進んでいるが、Peters異常に対する遺伝子治療はまだ実現していない4)。Peters異常の原因遺伝子が多様であること、発生期の一過性の異常に起因することから、出生後の遺伝子治療による角膜形態の回復は技術的に困難である。将来的には、出生前診断と組み合わせた早期介入の可能性が検討されている。